http://www.gacetamedica.com/gacetamedica/articulo.asp?vd=24/04/2005%2018:00:00&vh=01/05/2005%2018:00:00&num=&filtro=yes&hemeroteca=&idcat=706&idart=514452
Investigación: Estas técnicas serán asumidas en España de forma irremediable.
La genómica se generalizará en la clínica en una década
El grupo Euroespes presenta la primera Guía Mundial de Farmacogenómica.
Más de 2.000 familias en España ya están utilizando la tarjeta farmacogenética
Ramón Cacabelos, presidente de la Asociación Mundial de Medicina Genómica, presentó la primera Guía Mundial de Farmacogenómica en el marco del encuentro anual de Euroespes.
LA CORUÑA Viernes, 17 de Diciembre de 2010. Irene fernández
Su desarrollo y su descubrimiento son recientes, y la consolidación de su investigación aún se encuentra en fases tempranas, pero la medicina genómica en España y en el mundo poco a poco se acercará a la práctica.
"Aún estamos poniendo los cimientos, pero empezaremos a ver aspectos prácticos concretos de la medicina genómica en todos los campos de la medicina en una década", afirmó Ramón Cacabelos, presidente de la Asociación Mundial de Medicina Genómica, en el marco de la 5ª Conferencia Anual del grupo EuroEspes y 2ª Reunión Anual de la Asociación Mundial de Medicina Genómica, celebrada la semana pasada en La Coruña.
Y es que, los próximos 10 años marcarán la generalización de las técnicas genómicas, que serán "asumidas irremediablemente".
"Es un proceso imparable, es la punta de lanza de lo que tiene que ser el desarrollo biomédico en los próximos 50 años", continuó.
Muestra de un cimiento más para la extensión práctica de esta medicina ha sido el lanzamiento en el marco de esta conferencia de la primera Guía Mundial de Farmacogenómica, un proyecto en el que han participado más de 30 científicos internacionales y que ha sido coordinada por el doctor Cacabelos, del equipo de Euroespes.
Este centro médico se erige como referente internacional y está a la vanguardia de la investigación genómica, sobre todo en el área del sistema nervioso central.
"La guía es el primer ladrillo que se pone en la construcción internacional de la aplicación práctica de la farmacogenómica", dijo.
En total, el volumen, de más de 3.000 páginas que se presentará en forma de libro, CD y base de datos, recoge los 1.500 fármacos de mayor uso en todo el mundo presentados por su nombre químico y por más de 30.000 nombres comerciales.
Además, contiene información sobre los 600 genes más importantes en el metabolismo de los fármacos y los 4.000 más importantes en enfermedades humanas.
"En estos momentos, es lo más grande y lo único que hay en el mundo farmacogenómico, y nos ha llevado su realización cinco años", sostuvo.
En la actualidad, Euroespes cuenta con 6 megaproyectos de investigación.
Entre los más importantes, uno ha derivado en esta guía mundial gracias a la base de datos farmacogenómica, y el otro ya ha finalizado y está extendiéndose: la tarjeta farmacogenómica.
"Ya hay más de 2.000 familias en España que la usan, llevando así su genoma con ellos y los genes que influyen en su metabolismo de los fármacos", anunció.
En un futuro se universalizará.
De momento, países asiáticos y de América Latina la incorporarán próximamente, según Cacabelos.
Pero los aspectos éticos y legales de la medicina genómica no deben descuidarse.
Éste fue otro de los puntos de debate de la conferencia, en la que se concluyó que el uso de la tarjeta farmacogenómica debe ser siempre ultraconfidencial, y nunca esta información podrá servir para diferenciar o discriminar ni en detrimento de las personas, sino en beneficio.
Silenciamiento genético.
Máxime de la investigación genética es hoy la tecnología del silenciamiento genético o también llamado RNA de interferencia.
"Es una de las estrategias terapéuticas más poderosas en estos momentos porque nos ayuda a silenciar la expresión de un gen para que un cáncer no se manifieste o anular el crecimiento tumoral", explicó.
Aunque no es el único avance.
El desarrollo de bioproductos también está siendo determinante.
Entre ellos destacan 2:
a.- una lipoproteína de origen marino potente para reducir el colesterol como las estatinas y que lleva ya varios años en el mercado;
b.- y un inmunopotenciador que se utiliza fundamentalmente para enfermedades alérgicas, inflamatorias crónicas y para potenciar la inmunidad en el sida.
La característica distintiva de estos en concreto se distingue por que en función del genoma individual ejercen un efecto positivo, neutro o nulo.
Por tanto, "con cualquier bioproducto la optimización de la respuesta va a depender el perfil genómico de cada persona", aseveró.
miércoles, 22 de diciembre de 2010
sábado, 18 de diciembre de 2010
La Fundación Grünenthal y la investigación sobre el Dolor
http://www.actasanitaria.com/actasanitaria/frontend/desarrollo_noticia.jsp?idCanal=7&idContenido=22414
LA FUNDACIÓN GRÜNENTHAL CONVOCA EL PREMIO A LA MEJOR INVESTIGACIÓN EN DOLOR 2010
Madrid 04/11/2010
La Fundación Grünenthal convoca el Premio a la "Investigación en Dolor 201o" en colaboración con la Cátedra Extraordinaria del Dolor "Fundación Grünenthal" de la Universidad de Salamanca, con el objetivo de fomentar y reconocer la investigación científica en el campo del dolor, tanto en el ámbito epidemiológico, como experimental, farmacológico o clínico.
El premio está dotado con 6.000 euros y va dirigido a trabajos originales, de carácter independiente, publicados o aceptados para su publicación en revistas científicas desde el 1 de enero hasta el 1 de diciembre de 2010.
Las obras pueden ser presentadas por sus propios autores o por personas que no hayan participado en su elaboración.
El jurado calificador estará formado por el rector de la Universidad de Salamanca, el presidente del Patronato de la Fundación Grünenthal, el director de la Cátedra Extraordinaria del Dolor "Fundación Grünenthal" de la Universidad de Salamanca, así como por representantes de distintas sociedades científicas relevantes.
Bases de la ConvocatoriaLos candidatos interesados deben presentar los trabajos, por duplicado, a la Fundación Grünenthal (c/ Doctor Zamenhof, nº 36, 28027 de Madrid), o bien al correo electrónico fundacion.grunenthal@grunenthal.com, con la acreditación de publicación necesaria. La fecha límite de recepción de candidaturas es el 1 de diciembre de 2010.
LA FUNDACIÓN GRÜNENTHAL CONVOCA EL PREMIO A LA MEJOR INVESTIGACIÓN EN DOLOR 2010
Madrid 04/11/2010
La Fundación Grünenthal convoca el Premio a la "Investigación en Dolor 201o" en colaboración con la Cátedra Extraordinaria del Dolor "Fundación Grünenthal" de la Universidad de Salamanca, con el objetivo de fomentar y reconocer la investigación científica en el campo del dolor, tanto en el ámbito epidemiológico, como experimental, farmacológico o clínico.
El premio está dotado con 6.000 euros y va dirigido a trabajos originales, de carácter independiente, publicados o aceptados para su publicación en revistas científicas desde el 1 de enero hasta el 1 de diciembre de 2010.
Las obras pueden ser presentadas por sus propios autores o por personas que no hayan participado en su elaboración.
El jurado calificador estará formado por el rector de la Universidad de Salamanca, el presidente del Patronato de la Fundación Grünenthal, el director de la Cátedra Extraordinaria del Dolor "Fundación Grünenthal" de la Universidad de Salamanca, así como por representantes de distintas sociedades científicas relevantes.
Bases de la ConvocatoriaLos candidatos interesados deben presentar los trabajos, por duplicado, a la Fundación Grünenthal (c/ Doctor Zamenhof, nº 36, 28027 de Madrid), o bien al correo electrónico fundacion.grunenthal@grunenthal.com, con la acreditación de publicación necesaria. La fecha límite de recepción de candidaturas es el 1 de diciembre de 2010.
Genetica y pacientes con talla baja
http://www.actasanitaria.com/actasanitaria/frontend/desarrollo_noticia.jsp?idCanal=7&idContenido=22415
LOS HALLAZGOS GENÉTICOS APROXIMAN AL TRATAMIENTO PERSONALIZADO A PACIENTES CON TALLA BAJA
Valencia 04/11/2010
El tratamiento con hormona de crecimiento (GH) consigue que los pacientes con talla baja mejoren su velocidad de crecimiento al año de haber iniciado su administración.
Además, la talla continúa aumentando a un ritmo acelerado a lo largo de los 4 años siguientes al comienzo del tratamiento.
Así lo han demostrado los resultados del estudio GeNeSIS, presentados en Valencia.
El encuentro congregó a destacados especialistas en Endocrinología Pediátrica del panorama nacional para evaluar y compartir la evolución del tratamiento con GH desde la puesta en marcha de este estudio, hace 10 años.
El estudio y la reunión están patrocinados por Lilly.
El estudio GeNeSIS, realizado entre más de 17.000 pacientes procedentes de 30 países, se puso en marcha en 1999 como un estudio observacional a largo plazo sobre el tratamiento de la talla baja de diferentes causas, incluyendo el déficit de hormona de crecimiento.
"El principal objetivo de este estudio es determinar la seguridad y efectividad del tratamiento con hormona de crecimiento según las prácticas clínicas habituales de las consultas de endocrinología. Su valor fundamental es que tanto el manejo clínico de los pacientes como la recopilación de los datos se realizan exclusivamente bajo el único criterio de los médicos participantes", explica el Dr. Luis-Emilio García, del Departamento de Investigación Clínica de Lilly.
Y concluye que "a partir de estos resultados, se podrá desarrollar un modelo de predicción del crecimiento que ayudará, en gran medida, a identificar mejor a los pacientes que pueden tener una mayor respuesta al tratamiento. Esto puede facilitar la personalización de los tratamientos con hormona de crecimiento".
Por su parte, el Profesor Manuel Pombo, jefe de Servicio de Pediatría y de la Unidad de Endocrinología Pediátrica, Crecimiento y Adolescencia del Hospital Clínico Universitario de Santiago de Compostela, comentó:
"además de ofrecer datos de seguridad y efectividad de la GH, el estudio GeNeSIS ha permitido alcanzar hallazgos genéticos importantes, como caracterizar mejor las alteraciones del gen SHOX como responsable de algún tipo de trastorno de crecimiento".
El encuentro de Valencia también ha servido a los especialistas de la Endocrinología Pediátrica de nuestro país para hacer un recorrido por los avances logrados en la última década y valorar el posicionamiento de España a nivel mundial.
En este sentido, el Dr. Antonio Carrascosa, jefe del Servicio de Pediatría del Hospital Universitari Vall d’Hebron de Barcelona, reconoce que "el conocimiento sobre el tratamiento de la talla baja y el déficit de hormona de crecimiento ha mejorado mucho en los últimos años. En lo que a España se refiere podríamos decir que nuestro país está a niveles similares a los del resto de Europa".
LOS HALLAZGOS GENÉTICOS APROXIMAN AL TRATAMIENTO PERSONALIZADO A PACIENTES CON TALLA BAJA
Valencia 04/11/2010
El tratamiento con hormona de crecimiento (GH) consigue que los pacientes con talla baja mejoren su velocidad de crecimiento al año de haber iniciado su administración.
Además, la talla continúa aumentando a un ritmo acelerado a lo largo de los 4 años siguientes al comienzo del tratamiento.
Así lo han demostrado los resultados del estudio GeNeSIS, presentados en Valencia.
El encuentro congregó a destacados especialistas en Endocrinología Pediátrica del panorama nacional para evaluar y compartir la evolución del tratamiento con GH desde la puesta en marcha de este estudio, hace 10 años.
El estudio y la reunión están patrocinados por Lilly.
El estudio GeNeSIS, realizado entre más de 17.000 pacientes procedentes de 30 países, se puso en marcha en 1999 como un estudio observacional a largo plazo sobre el tratamiento de la talla baja de diferentes causas, incluyendo el déficit de hormona de crecimiento.
"El principal objetivo de este estudio es determinar la seguridad y efectividad del tratamiento con hormona de crecimiento según las prácticas clínicas habituales de las consultas de endocrinología. Su valor fundamental es que tanto el manejo clínico de los pacientes como la recopilación de los datos se realizan exclusivamente bajo el único criterio de los médicos participantes", explica el Dr. Luis-Emilio García, del Departamento de Investigación Clínica de Lilly.
Y concluye que "a partir de estos resultados, se podrá desarrollar un modelo de predicción del crecimiento que ayudará, en gran medida, a identificar mejor a los pacientes que pueden tener una mayor respuesta al tratamiento. Esto puede facilitar la personalización de los tratamientos con hormona de crecimiento".
Por su parte, el Profesor Manuel Pombo, jefe de Servicio de Pediatría y de la Unidad de Endocrinología Pediátrica, Crecimiento y Adolescencia del Hospital Clínico Universitario de Santiago de Compostela, comentó:
"además de ofrecer datos de seguridad y efectividad de la GH, el estudio GeNeSIS ha permitido alcanzar hallazgos genéticos importantes, como caracterizar mejor las alteraciones del gen SHOX como responsable de algún tipo de trastorno de crecimiento".
El encuentro de Valencia también ha servido a los especialistas de la Endocrinología Pediátrica de nuestro país para hacer un recorrido por los avances logrados en la última década y valorar el posicionamiento de España a nivel mundial.
En este sentido, el Dr. Antonio Carrascosa, jefe del Servicio de Pediatría del Hospital Universitari Vall d’Hebron de Barcelona, reconoce que "el conocimiento sobre el tratamiento de la talla baja y el déficit de hormona de crecimiento ha mejorado mucho en los últimos años. En lo que a España se refiere podríamos decir que nuestro país está a niveles similares a los del resto de Europa".
Europlan y las EE.RR.
http://www.actasanitaria.com/actasanitaria/frontend/desarrollo_noticia.jsp?idCanal=3&idContenido=22383
LA CONFERENCIA EUROPLAN ANALIZA LAS NECESIDADES DE LOS PACIENTES CON ENFERMEDADES RARAS
Madrid 03/11/2010
La Federación Española de Enfermedades Raras (FEDER) va a celebrar la Conferencia EUROPLAN, la mayor conferencia europea sobre patologías poco frecuentes, que tiene el objetivo de medir, a través de indicadores comunitarios, si la Estrategia Nacional destinada a estos pacientes alcanza los objetivos establecidos por la Comisión Europea en 2009.
FEDER organiza en España, los días 5 y 6 de noviembre, este encuentro, como la culminación de un proyecto europeo con el que se pretende crear un Documento Final de Conclusiones donde se trasladarán propuestas concretas para la Estrategia Nacional de Enfermedades Raras.
El objetivo es la transferencia de la Recomendación Europea a la Estrategia española y en ello han participado más de 120 expertos de todos los ámbitos.
Bajo la "lupa europea", este plan pretende garantizar la transferencia de la recomendación europea en la Estrategia Nacional de ER, así como identificar las buenas prácticas que se están llevando a cabo en nuestro país en el abordaje de estas patologías, tal y como explicó Rosa Sánchez de Vega, vicepresidenta de la Organización Europea de Enfermedades Raras (Eurordis).
Todo ello, se hará en el marco del proyecto internacional más importante en enfermedades raras, EUROPLAN, impulsado por la Organización Europea de Enfermedades Raras (EURORDIS), organizado por FEDER y coorganizado por el Centro de Referencia Estatal de Atención a Personas con Enfermedades Raras y sus Familias, dependiente del IMSERSO (CREER).
Muchas cuestiones por responder
EUROPLAN es un proyecto con indicadores orientado a resultados.
Sánchez de Vega aseguró que no se basa en una declaración de principios, ni un documento de buenas intenciones, pues, tras EUROPLAN se desarrollará un documento final de conclusiones donde se trasladarán propuestas concretas y directrices claras para implementar en la Estrategia Nacional de Enfermedades Raras.
Para lograr este documento final de conclusiones, más de 120 expertos de primera línea llevan más de 2 meses trabajando.
Se trata de investigadores, científicos, profesionales sociosanitarios, industria farmacéutica, Administración Nacional y Autonómica, plataformas sociales y representantes de los pacientes.
LA CONFERENCIA EUROPLAN ANALIZA LAS NECESIDADES DE LOS PACIENTES CON ENFERMEDADES RARAS
Madrid 03/11/2010
La Federación Española de Enfermedades Raras (FEDER) va a celebrar la Conferencia EUROPLAN, la mayor conferencia europea sobre patologías poco frecuentes, que tiene el objetivo de medir, a través de indicadores comunitarios, si la Estrategia Nacional destinada a estos pacientes alcanza los objetivos establecidos por la Comisión Europea en 2009.
FEDER organiza en España, los días 5 y 6 de noviembre, este encuentro, como la culminación de un proyecto europeo con el que se pretende crear un Documento Final de Conclusiones donde se trasladarán propuestas concretas para la Estrategia Nacional de Enfermedades Raras.
El objetivo es la transferencia de la Recomendación Europea a la Estrategia española y en ello han participado más de 120 expertos de todos los ámbitos.
Bajo la "lupa europea", este plan pretende garantizar la transferencia de la recomendación europea en la Estrategia Nacional de ER, así como identificar las buenas prácticas que se están llevando a cabo en nuestro país en el abordaje de estas patologías, tal y como explicó Rosa Sánchez de Vega, vicepresidenta de la Organización Europea de Enfermedades Raras (Eurordis).
Todo ello, se hará en el marco del proyecto internacional más importante en enfermedades raras, EUROPLAN, impulsado por la Organización Europea de Enfermedades Raras (EURORDIS), organizado por FEDER y coorganizado por el Centro de Referencia Estatal de Atención a Personas con Enfermedades Raras y sus Familias, dependiente del IMSERSO (CREER).
Muchas cuestiones por responder
EUROPLAN es un proyecto con indicadores orientado a resultados.
Sánchez de Vega aseguró que no se basa en una declaración de principios, ni un documento de buenas intenciones, pues, tras EUROPLAN se desarrollará un documento final de conclusiones donde se trasladarán propuestas concretas y directrices claras para implementar en la Estrategia Nacional de Enfermedades Raras.
Para lograr este documento final de conclusiones, más de 120 expertos de primera línea llevan más de 2 meses trabajando.
Se trata de investigadores, científicos, profesionales sociosanitarios, industria farmacéutica, Administración Nacional y Autonómica, plataformas sociales y representantes de los pacientes.
El Hospital 12 de Octubre investigaen EE.RR.
http://www.actasanitaria.com/actasanitaria/frontend/desarrollo_noticia.jsp?idCanal=3&idContenido=22352
EL HOSPITAL 12 DE OCTUBRE DEDICA MÁS DE 1 MILLÓN DE EUROS A INVESTIGAR ENFERMEDADES RARAS
Madrid 02/11/2010
Un equipo de 20 profesionales del Instituto de Investigación Hospital 12 de Octubre de la Comunidad de Madrid (i+12) desarrolla en la actualidad 11 proyectos de investigación centrados en enfermedades Raras mitocondriales y neuromusculares, con una dotación económica conjunta que supera 1 millón 200 mil euros. (1.200.000 €)
3 de los proyectos forman parte de la Acción Estratégica en Salud del Plan Nacional de I+D+i y tienen un financiación de casi 700.000 euros en total, incluyendo dotación de personal investigador.
** Uno está centrado en la investigación de los mecanismos moleculares y celulares que se ven alterados en las neuropatías mitocondriales infantiles y del adulto;
** otro en la identificación de las bases genéticas y fisiopatológicas de trastornos multisistémicos que producen un defecto enzimático mitocondrial y,
** un tercero, en el estudio de la función mitocondrial en ratones en un modelo de Esclerosis Lateral Amiotrófica (ELA).
Además de éstos, se desarrollan otros con fuentes de financiación pública dedicados también a la investigación en enfermedades mitocondriales y ELA, junto a varios ensayos clínicos independientes de la industria farmacéutica.
Este grupo de investigación cuenta con 2 laboratorios específicos en el Centro de Investigación Experimental del Instituto de Investigación Hospital 12 de Octubre (i+12).
Diagnóstico más rápido y eficaz
El Grupo i+12 participa también en el CIBER de Enfermedades Raras (CIBERER) y coordina un proyecto con el que se pretende estandarizar y aumentar el grado de automatización de metodologías que permitan un diagnóstico más rápido y eficaz en pacientes con sospecha de alternaciones del ADN mitocondrial, con el fin de transferir estas acciones a la cartera de servicios del Sistema Nacional de Salud.
Otros 2 proyectos, con financiación privada de la Fundación Mutua Madrileña y la Fundación Isabel Gemio, están dedicados respectivamente a la investigación de las mutaciones del ADN mitocondrial en encefalomiopatías mitocondriales y al estudio de terapias en fase preclínica de las distrofias musculares.
EL HOSPITAL 12 DE OCTUBRE DEDICA MÁS DE 1 MILLÓN DE EUROS A INVESTIGAR ENFERMEDADES RARAS
Madrid 02/11/2010
Un equipo de 20 profesionales del Instituto de Investigación Hospital 12 de Octubre de la Comunidad de Madrid (i+12) desarrolla en la actualidad 11 proyectos de investigación centrados en enfermedades Raras mitocondriales y neuromusculares, con una dotación económica conjunta que supera 1 millón 200 mil euros. (1.200.000 €)
3 de los proyectos forman parte de la Acción Estratégica en Salud del Plan Nacional de I+D+i y tienen un financiación de casi 700.000 euros en total, incluyendo dotación de personal investigador.
** Uno está centrado en la investigación de los mecanismos moleculares y celulares que se ven alterados en las neuropatías mitocondriales infantiles y del adulto;
** otro en la identificación de las bases genéticas y fisiopatológicas de trastornos multisistémicos que producen un defecto enzimático mitocondrial y,
** un tercero, en el estudio de la función mitocondrial en ratones en un modelo de Esclerosis Lateral Amiotrófica (ELA).
Además de éstos, se desarrollan otros con fuentes de financiación pública dedicados también a la investigación en enfermedades mitocondriales y ELA, junto a varios ensayos clínicos independientes de la industria farmacéutica.
Este grupo de investigación cuenta con 2 laboratorios específicos en el Centro de Investigación Experimental del Instituto de Investigación Hospital 12 de Octubre (i+12).
Diagnóstico más rápido y eficaz
El Grupo i+12 participa también en el CIBER de Enfermedades Raras (CIBERER) y coordina un proyecto con el que se pretende estandarizar y aumentar el grado de automatización de metodologías que permitan un diagnóstico más rápido y eficaz en pacientes con sospecha de alternaciones del ADN mitocondrial, con el fin de transferir estas acciones a la cartera de servicios del Sistema Nacional de Salud.
Otros 2 proyectos, con financiación privada de la Fundación Mutua Madrileña y la Fundación Isabel Gemio, están dedicados respectivamente a la investigación de las mutaciones del ADN mitocondrial en encefalomiopatías mitocondriales y al estudio de terapias en fase preclínica de las distrofias musculares.
Organizaciones del sector farmacéutico prestarán asistencia gratuita a pacientes con EE.RR.
http://www.actasanitaria.com/actasanitaria/frontend/desarrollo_noticia.jsp?idCanal=1&idContenido=22294
ORGANIZACIONES DEL SECTOR FARMACÉUTICO PRESTARÁN ASISTENCIA GRATUITA A PACIENTES CON ENFERMEDADES RARAS
Madrid 29/10/2010
* El Colegio de Farmacéuticos de Madrid (COFM),
* la distribuidora farmacéutica COFARES,
* la ONG Farmacéuticos sin Fronteras de España (FSFE),
* la Federación Española de Enfermedades Raras-Madrid (FEDER) y
* la Fundación FEDER para la investigación de Enfermedades Raras, con el apoyo de 11 laboratorios, han acordado desarrollar el proyecto 'En enfermedades raras sumamos todos'.
El acto de la firma, en la sede del Colegio Oficial de Farmacéuticos de Madrid, estuvo presidido por la Infanta Doña Elena.
Y se trata de un convenio de colaboración que prestará asistencia, a través de las oficinas de farmacia de la Comunidad de Madrid, a pacientes de enfermedades raras que cuentan con dificultades para acceder a los tratamientos no farmacológicos, ofreciéndoselos de manera gratuita.
FEDER será el interlocutor de referencia por su experiencia y contacto permanente con todas las asociaciones de afectados por enfermedades poco comunes;
la Fundación FEDER será la potenciadora del conocimiento científico es este campo, así como en el desarrollo de acciones de comunicación, formación y sensibilización; l
a ONG Farmacéuticos sin Fronteras de España aportará su experiencia en proyectos solidarios de ayuda sanitaria a colectivos necesitados;
COFARES pondrá a disposición la logística imprescindible para materializar envíos y entregas con la garantía y rapidez necesaria, y el Colegio de Farmacéuticos de Madrid (COFM) será el representante de todas las oficinas de farmacia de la Comunidad de Madrid que se quieran sumar voluntariamente al proyecto.
Todas estas entidades actuarán de forma totalmente altruista, así como los 11 laboratorios (La Roche Posay, BDF, Dermostética del Sur, Dentaid, OTC Ibérica, Pier Fabré-Avene-Ducray, Lacer, Galderma, Isdin y Nutrición Médica), que aportarán, igualmente sin coste, los productos demandados.
Un gasto elevado para los pacientes
Las Enfermedades raras engloban a un grupo de entre 5.000 y 8.000 patologías muy diversas, la mayoría de base genética, que afectan, de promedio, a uno por cada 2.000 habitantes.
Aunque los medicamentos de estos pacientes están cubiertos por el Sistema Nacional de Salud, la mayoría de estos enfermos requieren, además de dichos medicamentos, otros productos sanitarios (lentillas, cremas hidratantes, pomadas, geles, dentífricos y champús especiales, entre otros) que no están financiados por el sistema público, lo que supone para estos enfermos un gasto elevado que se une otros muchos cuidados.
El convenio, que ha tenido como punto de partida un estudio elaborado por la Federación de Enfermedades Raras sobre las necesidades socio sanitarias de los afectados por enfermedades poco frecuentes, ha servido como punto de partida y se espera que se vaya ampliando a toda la Comunidad de Madrid y al resto de España.
7 enfermedades raras
En principio, los beneficiarios son los pacientes afectados por:
+ Esclerodermia (piel dura),
+ Sjögren (afectación de glándulas exocrinas que dificulta las secreciones de las mucosas),
+ Mastocistosis (aumento de mastocititos que intervienen en las funciones que tienen que ver con la alergia),
+ Aniridia (aumento de la pupila que provoca fotofobia y otras alteraciones oftalmológicas),
+ Extrofia vesical (malformaciones de la vejiga y de la uretra),
+ Síndrome de Joubert (enfermedad neurológica que afecta a la coordinación de movimientos musculares), y
+ Enfermedad de Behçet (vasculitis con implicaciones oculares, neurológicas y arteriales).
ORGANIZACIONES DEL SECTOR FARMACÉUTICO PRESTARÁN ASISTENCIA GRATUITA A PACIENTES CON ENFERMEDADES RARAS
Madrid 29/10/2010
* El Colegio de Farmacéuticos de Madrid (COFM),
* la distribuidora farmacéutica COFARES,
* la ONG Farmacéuticos sin Fronteras de España (FSFE),
* la Federación Española de Enfermedades Raras-Madrid (FEDER) y
* la Fundación FEDER para la investigación de Enfermedades Raras, con el apoyo de 11 laboratorios, han acordado desarrollar el proyecto 'En enfermedades raras sumamos todos'.
El acto de la firma, en la sede del Colegio Oficial de Farmacéuticos de Madrid, estuvo presidido por la Infanta Doña Elena.
Y se trata de un convenio de colaboración que prestará asistencia, a través de las oficinas de farmacia de la Comunidad de Madrid, a pacientes de enfermedades raras que cuentan con dificultades para acceder a los tratamientos no farmacológicos, ofreciéndoselos de manera gratuita.
FEDER será el interlocutor de referencia por su experiencia y contacto permanente con todas las asociaciones de afectados por enfermedades poco comunes;
la Fundación FEDER será la potenciadora del conocimiento científico es este campo, así como en el desarrollo de acciones de comunicación, formación y sensibilización; l
a ONG Farmacéuticos sin Fronteras de España aportará su experiencia en proyectos solidarios de ayuda sanitaria a colectivos necesitados;
COFARES pondrá a disposición la logística imprescindible para materializar envíos y entregas con la garantía y rapidez necesaria, y el Colegio de Farmacéuticos de Madrid (COFM) será el representante de todas las oficinas de farmacia de la Comunidad de Madrid que se quieran sumar voluntariamente al proyecto.
Todas estas entidades actuarán de forma totalmente altruista, así como los 11 laboratorios (La Roche Posay, BDF, Dermostética del Sur, Dentaid, OTC Ibérica, Pier Fabré-Avene-Ducray, Lacer, Galderma, Isdin y Nutrición Médica), que aportarán, igualmente sin coste, los productos demandados.
Un gasto elevado para los pacientes
Las Enfermedades raras engloban a un grupo de entre 5.000 y 8.000 patologías muy diversas, la mayoría de base genética, que afectan, de promedio, a uno por cada 2.000 habitantes.
Aunque los medicamentos de estos pacientes están cubiertos por el Sistema Nacional de Salud, la mayoría de estos enfermos requieren, además de dichos medicamentos, otros productos sanitarios (lentillas, cremas hidratantes, pomadas, geles, dentífricos y champús especiales, entre otros) que no están financiados por el sistema público, lo que supone para estos enfermos un gasto elevado que se une otros muchos cuidados.
El convenio, que ha tenido como punto de partida un estudio elaborado por la Federación de Enfermedades Raras sobre las necesidades socio sanitarias de los afectados por enfermedades poco frecuentes, ha servido como punto de partida y se espera que se vaya ampliando a toda la Comunidad de Madrid y al resto de España.
7 enfermedades raras
En principio, los beneficiarios son los pacientes afectados por:
+ Esclerodermia (piel dura),
+ Sjögren (afectación de glándulas exocrinas que dificulta las secreciones de las mucosas),
+ Mastocistosis (aumento de mastocititos que intervienen en las funciones que tienen que ver con la alergia),
+ Aniridia (aumento de la pupila que provoca fotofobia y otras alteraciones oftalmológicas),
+ Extrofia vesical (malformaciones de la vejiga y de la uretra),
+ Síndrome de Joubert (enfermedad neurológica que afecta a la coordinación de movimientos musculares), y
+ Enfermedad de Behçet (vasculitis con implicaciones oculares, neurológicas y arteriales).
Fundacion Mehuer y las EE.RR
http://www.actasanitaria.com/actasanitaria/frontend/desarrollo_noticia.jsp?idCanal=24&idContenido=23132
EL COLEGIO DE FARMACÉUTICOS DE SEVILLA PROMUEVE LA FUNDACIÓN MEHUER PARA LA INVESTIGACIÓN Y EL TRATAMIENTO DE LAS ENFERMEDADES RARAS
Sevilla 09/12/2010
Este jueves se ha celebrado en el Palacio Arzobispal de Sevilla el acto de constitución del Patronato de la Fundación Mehuer, que promoverá desde Sevilla la investigación de terapias y tratamientos para las enfermedades raras, aquellas que tienen una baja frecuencia en la población y que son potencialmente mortales o debilitantes.
Esta Fundación nace promovida por el Colegio de Farmacéuticos de Sevilla, que ha logrado recabar en esta iniciativa el apoyo de empresarios y directivos algunas de las instituciones y empresas más representativas y con mayor liderazgo en la sociedad civil sevillana y andaluza.
Composición del PatronatoEl Presidente del Colegio de Farmacéuticos de Sevilla, Manuel Pérez Fernández, es el Presidente la Fundación Mehuer, cuyo Patronato está constituido además por los siguientes miembros: Juan José Asenjo Pelegrina, Arzobispo de Sevilla; Ricardo Abaurre Lorente, Director de Relaciones Institucionales de Abengoa Solar; Miguel Ángel Bermudo Valero, director de Asesoría Jurídica de Heineken España; Antonio García de Castro, director de la Fundación Instituto Internacinal San Telmo; Félix Hernández Callejas, Director General de Herba Rice Mills; Gonzalo Madariaga de Parias; Antonio Pulido Gutiérrez; presidente de Cajasol; y los siguientes miembros de la Junta de Gobierno del Colegio de Farmacéuticos de Sevilla: Manuel Ojeda Casares, Vicepresidente; Juan Pedro Vaquero Prada, Secretario; Juan Luis Barea Ledesma, Tesorero; María Isabel de Andrés Martín, Vicesecretaria; María Teresa Díaz Carmona, Secretaria Técnica; Leopoldo Gutiérrez-Alviz Conradi, Asesor Jurídico y Pedro Bueno López, Gerente.
Por qué una Fundación
En los últimos años se han logrado algunos avances importantes para mejorar la calidad de vida de los pacientes con enfermedades raras, sin embargo todo eso no es suficiente.
Sigue faltando una actividad más decidida en todos los terrenos que afectan a estas patologías y la conciencia de que estamos ante un problema de salud enormemente complejo que requiere de la implicación sucesiva permanente y continuada de toda la sociedad organismos nacionales, internacionales, estatales, autonómicos, sanitarios, sociales... y, sobre todo, de la sociedad civil.
Así lo vio el Real e Ilustre Colegio de Farmacéuticos de Sevilla hace más de 12 años, cuando decidió implicarse, con la inestimable colaboración del profesor Santiago Grisolía, en la mejora de las condiciones de vida de los pacientes con enfermedades raras y organizó, en el año 2000, el Iº Congreso Internacional de Medicamentos Huérfanos y Enfermedades Raras.
Y así lo sigue viendo hoy, después de haber organizado otros IV Congresos Internacionales y haber promovido numerosas iniciativas encaminadas a la misma finalidad: 7 becas de investigación, 3 premios periodísticos, la Declaración de Sevilla sobre Enfermedades Raras, así como numerosas participaciones en congresos nacionales e internacionales.
La Fundación Mehuer nace precisamente con el objetivo de reforzar esta actividad, implicando a otras empresas e instituciones líderes de la sociedad civil sevillana y andaluza, y con la intención de extender ese compromiso y esa sensibilidad entre otras muchas empresas y entidades, así como entre los propios ciudadanos.
EL COLEGIO DE FARMACÉUTICOS DE SEVILLA PROMUEVE LA FUNDACIÓN MEHUER PARA LA INVESTIGACIÓN Y EL TRATAMIENTO DE LAS ENFERMEDADES RARAS
Sevilla 09/12/2010
Este jueves se ha celebrado en el Palacio Arzobispal de Sevilla el acto de constitución del Patronato de la Fundación Mehuer, que promoverá desde Sevilla la investigación de terapias y tratamientos para las enfermedades raras, aquellas que tienen una baja frecuencia en la población y que son potencialmente mortales o debilitantes.
Esta Fundación nace promovida por el Colegio de Farmacéuticos de Sevilla, que ha logrado recabar en esta iniciativa el apoyo de empresarios y directivos algunas de las instituciones y empresas más representativas y con mayor liderazgo en la sociedad civil sevillana y andaluza.
Composición del PatronatoEl Presidente del Colegio de Farmacéuticos de Sevilla, Manuel Pérez Fernández, es el Presidente la Fundación Mehuer, cuyo Patronato está constituido además por los siguientes miembros: Juan José Asenjo Pelegrina, Arzobispo de Sevilla; Ricardo Abaurre Lorente, Director de Relaciones Institucionales de Abengoa Solar; Miguel Ángel Bermudo Valero, director de Asesoría Jurídica de Heineken España; Antonio García de Castro, director de la Fundación Instituto Internacinal San Telmo; Félix Hernández Callejas, Director General de Herba Rice Mills; Gonzalo Madariaga de Parias; Antonio Pulido Gutiérrez; presidente de Cajasol; y los siguientes miembros de la Junta de Gobierno del Colegio de Farmacéuticos de Sevilla: Manuel Ojeda Casares, Vicepresidente; Juan Pedro Vaquero Prada, Secretario; Juan Luis Barea Ledesma, Tesorero; María Isabel de Andrés Martín, Vicesecretaria; María Teresa Díaz Carmona, Secretaria Técnica; Leopoldo Gutiérrez-Alviz Conradi, Asesor Jurídico y Pedro Bueno López, Gerente.
Por qué una Fundación
En los últimos años se han logrado algunos avances importantes para mejorar la calidad de vida de los pacientes con enfermedades raras, sin embargo todo eso no es suficiente.
Sigue faltando una actividad más decidida en todos los terrenos que afectan a estas patologías y la conciencia de que estamos ante un problema de salud enormemente complejo que requiere de la implicación sucesiva permanente y continuada de toda la sociedad organismos nacionales, internacionales, estatales, autonómicos, sanitarios, sociales... y, sobre todo, de la sociedad civil.
Así lo vio el Real e Ilustre Colegio de Farmacéuticos de Sevilla hace más de 12 años, cuando decidió implicarse, con la inestimable colaboración del profesor Santiago Grisolía, en la mejora de las condiciones de vida de los pacientes con enfermedades raras y organizó, en el año 2000, el Iº Congreso Internacional de Medicamentos Huérfanos y Enfermedades Raras.
Y así lo sigue viendo hoy, después de haber organizado otros IV Congresos Internacionales y haber promovido numerosas iniciativas encaminadas a la misma finalidad: 7 becas de investigación, 3 premios periodísticos, la Declaración de Sevilla sobre Enfermedades Raras, así como numerosas participaciones en congresos nacionales e internacionales.
La Fundación Mehuer nace precisamente con el objetivo de reforzar esta actividad, implicando a otras empresas e instituciones líderes de la sociedad civil sevillana y andaluza, y con la intención de extender ese compromiso y esa sensibilidad entre otras muchas empresas y entidades, así como entre los propios ciudadanos.
domingo, 31 de octubre de 2010
Galicia: los Pediatras en pie de "Guerra"
http://www.cincodias.com/articulo/economia/medico-atencion-primaria-reclama-libre-acceso-internet/20101025cdscdieco_8/cdseco/#
El médico de atención primaria reclama libre acceso a internet
Pediatras y facultativos de familia denuncian que 8 regiones limitan su uso.
Andalucía, Valencia, Galicia y La Rioja ponen "restricciones fuertes" de acceso.
Natalia Sanmartin Fenollera - Madrid - 25/10/2010
En Galicia el médico es un presunto delincuente.
No se puede llamar a móviles, internet está totalmente mutilado, prácticamente la única página importante a la que se puede acceder desde aquí libremente es www.Fisterra.com.
Quizás por eso estamos en los confines de la tierra".
Así de explícita se muestra una de las facultativas que apoyan en la red la campaña Internet en la consulta: una necesidad, que ha puesto en marcha el colectivo de pediatras de atención primaria para reivindicar el derecho de los médicos a acceder sin cortapisas a internet desde la consulta.
Sin embargo, no es un problema que ataña sólo a los facultativos gallegos.
Según los promotores de la iniciativa, que está apoyada por:
1.- la Asociación Española de Pediatría,
2.-Sociedad Española de Medicina de Familia y Comunitaria,
3.-Sociedad Española de Médicos de Atención Primaria y
4.-Sociedad Española de Médicos Generales y de Familia,
Hay 4 comunidades autónomas donde los médicos tienen "restricciones fuertes" para acceder a internet: Galicia, Andalucía, Comunidad Valenciana y La Rioja.
A ellas hay que sumar otras 4: -Extremadura, Castilla-La Mancha, Murcia y País Vasco- que cuentan con "restricciones suaves".
Los datos provienen de un sondeo realizado por la Asociación Española de Pediatría de Atención Primaria entre los responsables de sus asociaciones autonómicas federadas.
"Resulta evidente que esta actitud de censura es precisamente una muestra de desconocimiento, tanto de la utilidad como del funcionamiento de esas nuevas (ya no tanto) tecnologías y además constituye una flagrante desconsideración hacia el médico y sus pacientes", sostienen las sociedades médicas que impulsan la denuncia.
Así, los médicos que soportan restricciones en el acceso a la red no sólo se ven privados de una fuente de información profesional fundamental para su trabajo, sino de los recursos de formación científica online accesibles a través de internet, de la posibilidad de comunicarse con compañeros, pacientes y gestores y de las iniciativas de participación que hoy en día ofrece la red para comunicar a médicos y colectivos de pacientes.
"La situación actual en la que se encuentra la conexión a internet en las consultas de atención primaria andaluzas sería similar a lo que ocurriría si alguien, a hurtadillas, hubiese entrado en nuestra consulta y nos hubiese dejado en un cajón escondido, cerrado con un candado (que en realidad no funciona y se abre tirando de él), un equipo obsoleto sin manual de instrucciones que nos conecta (si sabemos) a algunas páginas web", describe gráficamente el pediatra Rafael Jiménez Alés.
El facultativo recomienda con sorna a sus compañeros de profesión que no olviden "que siempre hay un colegio cerca en el que pedir prestado un portátil con internet a un alumno. Bastará que le expidas un certificado de la solicitud de préstamo".
La sanidad gallega anuncia mejoras
La dirección de Asistencia Sanitaria gallega escribió el pasado 15 de octubre una carta a los facultativos de la región para anunciarles una próxima mejora de las condiciones de acceso a internet.
Según se reconoce en la carta, en Galicia sólo 57 centros de salud de los 440 que cubren al 98% de la población gallega disponen de "líneas de telecomunicaciones de alta capacidad", y será a esos centros a los que se les dotará de mayor "flexibilidad" de acceso.
"Iremos introduciendo nuevos centros a medida que se vayan ampliando las líneas de comunicaciones de los mismos", se anuncia en la misiva, además de explicar, entre otras novedades, que antes de final de año se instalará "una nueva intranet en la que se facilite la búsqueda de información".
El médico de atención primaria reclama libre acceso a internet
Pediatras y facultativos de familia denuncian que 8 regiones limitan su uso.
Andalucía, Valencia, Galicia y La Rioja ponen "restricciones fuertes" de acceso.
Natalia Sanmartin Fenollera - Madrid - 25/10/2010
En Galicia el médico es un presunto delincuente.
No se puede llamar a móviles, internet está totalmente mutilado, prácticamente la única página importante a la que se puede acceder desde aquí libremente es www.Fisterra.com.
Quizás por eso estamos en los confines de la tierra".
Así de explícita se muestra una de las facultativas que apoyan en la red la campaña Internet en la consulta: una necesidad, que ha puesto en marcha el colectivo de pediatras de atención primaria para reivindicar el derecho de los médicos a acceder sin cortapisas a internet desde la consulta.
Sin embargo, no es un problema que ataña sólo a los facultativos gallegos.
Según los promotores de la iniciativa, que está apoyada por:
1.- la Asociación Española de Pediatría,
2.-Sociedad Española de Medicina de Familia y Comunitaria,
3.-Sociedad Española de Médicos de Atención Primaria y
4.-Sociedad Española de Médicos Generales y de Familia,
Hay 4 comunidades autónomas donde los médicos tienen "restricciones fuertes" para acceder a internet: Galicia, Andalucía, Comunidad Valenciana y La Rioja.
A ellas hay que sumar otras 4: -Extremadura, Castilla-La Mancha, Murcia y País Vasco- que cuentan con "restricciones suaves".
Los datos provienen de un sondeo realizado por la Asociación Española de Pediatría de Atención Primaria entre los responsables de sus asociaciones autonómicas federadas.
"Resulta evidente que esta actitud de censura es precisamente una muestra de desconocimiento, tanto de la utilidad como del funcionamiento de esas nuevas (ya no tanto) tecnologías y además constituye una flagrante desconsideración hacia el médico y sus pacientes", sostienen las sociedades médicas que impulsan la denuncia.
Así, los médicos que soportan restricciones en el acceso a la red no sólo se ven privados de una fuente de información profesional fundamental para su trabajo, sino de los recursos de formación científica online accesibles a través de internet, de la posibilidad de comunicarse con compañeros, pacientes y gestores y de las iniciativas de participación que hoy en día ofrece la red para comunicar a médicos y colectivos de pacientes.
"La situación actual en la que se encuentra la conexión a internet en las consultas de atención primaria andaluzas sería similar a lo que ocurriría si alguien, a hurtadillas, hubiese entrado en nuestra consulta y nos hubiese dejado en un cajón escondido, cerrado con un candado (que en realidad no funciona y se abre tirando de él), un equipo obsoleto sin manual de instrucciones que nos conecta (si sabemos) a algunas páginas web", describe gráficamente el pediatra Rafael Jiménez Alés.
El facultativo recomienda con sorna a sus compañeros de profesión que no olviden "que siempre hay un colegio cerca en el que pedir prestado un portátil con internet a un alumno. Bastará que le expidas un certificado de la solicitud de préstamo".
La sanidad gallega anuncia mejoras
La dirección de Asistencia Sanitaria gallega escribió el pasado 15 de octubre una carta a los facultativos de la región para anunciarles una próxima mejora de las condiciones de acceso a internet.
Según se reconoce en la carta, en Galicia sólo 57 centros de salud de los 440 que cubren al 98% de la población gallega disponen de "líneas de telecomunicaciones de alta capacidad", y será a esos centros a los que se les dotará de mayor "flexibilidad" de acceso.
"Iremos introduciendo nuevos centros a medida que se vayan ampliando las líneas de comunicaciones de los mismos", se anuncia en la misiva, además de explicar, entre otras novedades, que antes de final de año se instalará "una nueva intranet en la que se facilite la búsqueda de información".
Fundación BBVA y la Investigación sobre el Cáncer
http://www.cincodias.com/articulo/economia/Fundacion-BBVA-reune-elite-investigadores-cancer/20101025cdscdieco_12/cdseco/
Simposio internacional: Fundación BBVA reúne a la élite de los investigadores del cáncer
Cinco Días - Madrid - 25/10/2010
El CNIO de Biología Celular, la Fundación BBVA y el Grupo Editorial Nature reúnen hasta el próximo miércoles, 27 de octubre, a los líderes mundiales en la investigación que marcará el tratamiento del cáncer en la próxima década.
El simposio, denominado Fronteras en la progresión tumoral, se está celebrando en Madrid con la participación de Joan Massagué, director del Programa de Biología y Genética del Cáncer del Sloan-Kettering Institute, quien ha manifestado que "los ponentes y participantes en este encuentro expondrán nuevos hallazgos respecto a los genes y proteínas que las células tumorales utilizan para llevar a cabo la metástasis".
Entre ellos, se encuentran Michael Karin, distinguished professor de Farmacología en la Universidad de California San Diego, y Zena Werb, catedrática de Anatomía en la Universidad de California en San Francisco, que expondrá sus descubrimientos este lunes por la tarde.
También participa Erwin Wagner, director del Programa Fundación BBVA-CNIO de Biología Celular del Cáncer.
Por su parte, Douglas Hanahan, director del Instituto Suizo para la Investigación Experimental en Cáncer en la École Polytechnique Fédérale de Lausanne lo hará el miércoles, clausurando el simposio.
En total, 32 ponentes de diferentes especialidades y nacionalidades que abordan los principales aspectos de esta enfermedad.
El encuentro se articulará en torno a 5 sesiones:
1.-Genética, modelos animales y mecanismos;
2.-La célula cancerosa metastásica;
3.-Moduladores inflamatorios;
4.-Terapias antimetastásicas basadas en mecanismos, y
5.-Angiogénesis y terapia antiangiogénica.
Según Massagué, "hoy nos estamos beneficiando de tratamientos contra el cáncer que son el fruto de la investigación básica llevada a cabo hace 10 o más años. El simposio se centrará en la investigación básica que está poniendo las bases para los avances terapéuticos de la próxima década. Hoy estamos en condiciones de investigar el cáncer no como un tumor de un órgano determinado, sino como una enfermedad sistémica que afecta a todo el organismo".
Michael Karin inaugurará la conferencia
La conferencia inaugural será impartida por Michael Karin, quien en 2007 estableció el vínculo entre inflamación y cáncer.
2 de sus estudios demuestran que la obesidad es un factor promotor de tumores y que la exposición al tabaco aumenta el tamaño y la difusión de los tumores en el pulmón en ratones con cáncer, fenómenos que se explican también por la inflamación.
Karin expondrá hallazgos recientes, como el de que "varias moléculas cuya inhibición puede impedir o atenuar la metástasis en ratones, puede tener un efecto similar en el cáncer de próstata y de mama en humanos".
Simposio internacional: Fundación BBVA reúne a la élite de los investigadores del cáncer
Cinco Días - Madrid - 25/10/2010
El CNIO de Biología Celular, la Fundación BBVA y el Grupo Editorial Nature reúnen hasta el próximo miércoles, 27 de octubre, a los líderes mundiales en la investigación que marcará el tratamiento del cáncer en la próxima década.
El simposio, denominado Fronteras en la progresión tumoral, se está celebrando en Madrid con la participación de Joan Massagué, director del Programa de Biología y Genética del Cáncer del Sloan-Kettering Institute, quien ha manifestado que "los ponentes y participantes en este encuentro expondrán nuevos hallazgos respecto a los genes y proteínas que las células tumorales utilizan para llevar a cabo la metástasis".
Entre ellos, se encuentran Michael Karin, distinguished professor de Farmacología en la Universidad de California San Diego, y Zena Werb, catedrática de Anatomía en la Universidad de California en San Francisco, que expondrá sus descubrimientos este lunes por la tarde.
También participa Erwin Wagner, director del Programa Fundación BBVA-CNIO de Biología Celular del Cáncer.
Por su parte, Douglas Hanahan, director del Instituto Suizo para la Investigación Experimental en Cáncer en la École Polytechnique Fédérale de Lausanne lo hará el miércoles, clausurando el simposio.
En total, 32 ponentes de diferentes especialidades y nacionalidades que abordan los principales aspectos de esta enfermedad.
El encuentro se articulará en torno a 5 sesiones:
1.-Genética, modelos animales y mecanismos;
2.-La célula cancerosa metastásica;
3.-Moduladores inflamatorios;
4.-Terapias antimetastásicas basadas en mecanismos, y
5.-Angiogénesis y terapia antiangiogénica.
Según Massagué, "hoy nos estamos beneficiando de tratamientos contra el cáncer que son el fruto de la investigación básica llevada a cabo hace 10 o más años. El simposio se centrará en la investigación básica que está poniendo las bases para los avances terapéuticos de la próxima década. Hoy estamos en condiciones de investigar el cáncer no como un tumor de un órgano determinado, sino como una enfermedad sistémica que afecta a todo el organismo".
Michael Karin inaugurará la conferencia
La conferencia inaugural será impartida por Michael Karin, quien en 2007 estableció el vínculo entre inflamación y cáncer.
2 de sus estudios demuestran que la obesidad es un factor promotor de tumores y que la exposición al tabaco aumenta el tamaño y la difusión de los tumores en el pulmón en ratones con cáncer, fenómenos que se explican también por la inflamación.
Karin expondrá hallazgos recientes, como el de que "varias moléculas cuya inhibición puede impedir o atenuar la metástasis en ratones, puede tener un efecto similar en el cáncer de próstata y de mama en humanos".
Guia Metabólica sobre Osteocondromas (Exostosis Múltiples)
http://www.guiametabolica.org/informacion/exostosis-multiple-hereditaria
El Hospital San Juan de Dios (L´Hospital Sant Joan de Déu) - Barcelona- ha publicado una Guia Metabólica sobre las Exostosis Multiples u Osteocondromas Multiples Congénitos.
Pinchar el enlace y teneis la información: http://www.guiametabolica.org/category/tags/osteocondroma
Asociación Española de Osteocondromas Múltiples Congénitos (AEOMC)
El Hospital San Juan de Dios (L´Hospital Sant Joan de Déu) - Barcelona- ha publicado una Guia Metabólica sobre las Exostosis Multiples u Osteocondromas Multiples Congénitos.
Pinchar el enlace y teneis la información: http://www.guiametabolica.org/category/tags/osteocondroma
Asociación Española de Osteocondromas Múltiples Congénitos (AEOMC)
Barcelona: Seminario Internacional de Ortopedia Infantil
http://www.hsjdbcn.org/portal/ca/web/2149152853
XV Seminari Internacional d’Ortopèdia Infantil
08-09-2010
Els propers dies 5 i 6 de novembre se celebrarà a Barcelona el XVè Seminari Internacional d’Ortopèdia Infantil que comptarà amb la intervenció de diversos especialistes del nostre Hospital i d’altres experts provinents de països com Singapur, Estats Units, Argentina i França, entre d'altres.
La trobada està dirigida pel Dr. Ramon Huguet, cap de Cirurgia Ortopèdica i Traumatologia de l’Hospital Sant Joan de Déu i el Dr. Julio de Pablos, cirurgià ortopèdic de l’Hospital de Navarra i de l’Hospital San Juan de Dios de Pamplona .
La inscripció es pot fer a través de la web: [ http://www.depablos-bruguera.com/ ]
[ Fulletó del seminari internacional d'ortopèdia infantil (pdf) ]
Hospital Sant Joan de Déu
XV Seminari Internacional d’Ortopèdia Infantil
08-09-2010
Els propers dies 5 i 6 de novembre se celebrarà a Barcelona el XVè Seminari Internacional d’Ortopèdia Infantil que comptarà amb la intervenció de diversos especialistes del nostre Hospital i d’altres experts provinents de països com Singapur, Estats Units, Argentina i França, entre d'altres.
La trobada està dirigida pel Dr. Ramon Huguet, cap de Cirurgia Ortopèdica i Traumatologia de l’Hospital Sant Joan de Déu i el Dr. Julio de Pablos, cirurgià ortopèdic de l’Hospital de Navarra i de l’Hospital San Juan de Dios de Pamplona .
La inscripció es pot fer a través de la web: [ http://www.depablos-bruguera.com/ ]
[ Fulletó del seminari internacional d'ortopèdia infantil (pdf) ]
Hospital Sant Joan de Déu
miércoles, 20 de octubre de 2010
Los Osteocondromas, tumores benignos del hueso
http://www.datoanuncios.org/?a=28288
Lunes 18 de Octubre del año 2010
Tumores benignos del hueso:Los osteocondromas...
Los tumores óseos plazo, todos los tumores del tejido óseo se puede resumir.
Ellos son benignos y malignos dividido, con los tumores malignos, pero las caídas más importantes, las formas benignas son comunes.
Dependiendo de la naturaleza del tejido de la matriz del hueso, cartílago o de otro tipo de tumores se pueden distinguir:
1.- tumores osteogénica del tejido óseo base, por ejemplo, Osteoma, osteoma osteoide, osteoblastoma
2.- Condrogénica tumores del tejido del cartílago en la base, por ejemplo, Condroma condroblastoma, fibroma condromixoide, osteocondroma.
3.- Vasogénico tumores en la base de los buques, por ejemplo Hemangioma, linfangioma, tumores glómicos.
4.- Otros orígenes: por ejemplo, Lipoma (tejido adiposo), neurofibroma (tejido nervioso), tumor de células gigantes del hueso
Los 2 principales tumores óseos benignos son el osteocondroma y condroma.
Los osteocondromas
Ellos son los tumores óseos benignos más comunes y están compuestas de cartílago y tejido óseo. Puede ocurrir en los huesos individuales (solitario) o generalizada en todo el cuerpo (múltiple).
Esto se atribuye a los múltiples osteocondromas un componente hereditario.
Ocurren con mayor frecuencia en niños y en la niñez.
Los osteocondromas se siente en los huesos y partes cartilaginosas se realizan en la radiografía no plenamente representados.
Se producen principalmente en los puntos cercanos a las articulaciones de las extremidades y huesos de los dedos.
Frecuencia
Los tumores óseos benignos son mucho más comunes que malignas.
Este es el más común de tumor benigno de Osteocondroma condroblastoma y dar condromixoide. Conecte el contrario, con menos frecuencia.
Causas
Las razones para el desarrollo de los tumores óseos benignos son todavía en gran parte desconocida.
En casos raros, las bases genéticas juegan un papel importante.
Algunos tipos de cáncer son hereditarios rara vez se muestra, se incluyen osteocondromas.
Si los factores físicos, químicos o bioquímicos implicados en el desarrollo y aún no ha bekannt.Die causas para el desarrollo de los tumores óseos benignos son todavía en gran parte desconocida. En casos raros, las bases genéticas juegan un papel importante.
Algunos tipos de cáncer son hereditarios rara vez se muestra, se incluyen osteocondromas.
Ya sea física, química o bioquímica de factores están involucrados en el desarrollo no se conoce.
Los síntomas
Dependiendo del tipo de tumor, el asiento y diseminarse puede variar, los síntomas de los tumores óseos benignos. Casi todos los pacientes se quejan de dolor en las zonas afectadas.
A menudo, la deformación de las articulaciones o los huesos pueden ser observadas.
Y fracturas óseas causadas por el crecimiento del tumor se producen.
En la infancia puede llevar a problemas de crecimiento.
Los tumores óseos benignos se producen en los lugares preferidos en el cuerpo.
Las costillas son casi nunca cambia tumorales, mientras que por ejemplo, Los osteocondromas son más frecuentes en los puntos cercanos a las articulaciones de las extremidades, especialmente las piernas.
Diagnóstico
El diagnóstico presuntivo fue planteada por la historia (entrevista) con un cuestionario detallado acerca de los síntomas, el curso anterior de la historia de la enfermedad y la familia.
En el examen físico, el dolor y la deformación puede ser evidente.
Para confirmar el diagnóstico, una imagen de rayos X se produce en dos niveles.
Aquí, los cambios óseos típicos se observan.
En los resultados poco claros, por ejemplo sobre la cuestión de si se trata de tumores benignos o malignos, otros estudios como la tomografía computarizada (TC) y resonancia magnética (IRM) se llevan a cabo.
Si no es el diagnóstico sobre la base de la proyección de imagen para dejar en claro, una muestra (biopsia) se recomienda.
El diagnóstico presuntivo fue planteada por la historia (entrevista) con un cuestionario detallado acerca de los síntomas, el curso anterior de la historia de la enfermedad y la familia.
En el examen físico, el dolor y la deformación puede ser evidente.
Para confirmar el diagnóstico, una imagen de rayos X se produce en dos niveles.
Aquí, los cambios óseos típicos se observan.
En los resultados poco claros, por ejemplo sobre la cuestión de si se trata de tumores benignos o malignos, otros estudios como la tomografía computarizada (TC) y resonancia magnética (IRM) se llevan a cabo.
Si no es el diagnóstico sobre la base de la proyección de imagen para dejar en claro, una muestra (biopsia) se recomienda.
Terapia
Dependiendo del tipo de tumor y diseminarse puede variar el tratamiento.
Es, sin embargo, generalmente a partir de la extirpación quirúrgica del tumor.
Con poco tumores sintomáticos bajo la estrecha supervisión también puede ser una de esperar y ser ingeridos.
Fracturas de huesos se puede tratar mediante intervención quirúrgica o conservadora.
Los osteocondromas
El tratamiento quirúrgico se debe hacer si el tumor es sintomático.
Si hay un aumento de tamaño después de el crecimiento, por lo que la operación también se recomienda, como puede ocurrir en casos raros, un desarrollo de un tumor maligno.
Cuidado Posterior
El seguimiento se basa siempre en el tipo de tumor y con el médico a consultar.
Pronóstico
El pronóstico de los tumores óseos benignos generalmente es bueno.
Con el objetivo de la detección precoz de la recurrencia del tumor hasta los exámenes se recomiendan.
Sólo el Exostosenkrankheit tendencia familiar en algunos casos a convertirse en malignos.
Si en estos pacientes después de la finalización de crecimiento se producen un aumento en el tamaño de la exostosis, una nueva investigación / operación se recomienda.
El pronóstico es aún estima que será favorable.
Lunes 18 de Octubre del año 2010
Tumores benignos del hueso:Los osteocondromas...
Los tumores óseos plazo, todos los tumores del tejido óseo se puede resumir.
Ellos son benignos y malignos dividido, con los tumores malignos, pero las caídas más importantes, las formas benignas son comunes.
Dependiendo de la naturaleza del tejido de la matriz del hueso, cartílago o de otro tipo de tumores se pueden distinguir:
1.- tumores osteogénica del tejido óseo base, por ejemplo, Osteoma, osteoma osteoide, osteoblastoma
2.- Condrogénica tumores del tejido del cartílago en la base, por ejemplo, Condroma condroblastoma, fibroma condromixoide, osteocondroma.
3.- Vasogénico tumores en la base de los buques, por ejemplo Hemangioma, linfangioma, tumores glómicos.
4.- Otros orígenes: por ejemplo, Lipoma (tejido adiposo), neurofibroma (tejido nervioso), tumor de células gigantes del hueso
Los 2 principales tumores óseos benignos son el osteocondroma y condroma.
Los osteocondromas
Ellos son los tumores óseos benignos más comunes y están compuestas de cartílago y tejido óseo. Puede ocurrir en los huesos individuales (solitario) o generalizada en todo el cuerpo (múltiple).
Esto se atribuye a los múltiples osteocondromas un componente hereditario.
Ocurren con mayor frecuencia en niños y en la niñez.
Los osteocondromas se siente en los huesos y partes cartilaginosas se realizan en la radiografía no plenamente representados.
Se producen principalmente en los puntos cercanos a las articulaciones de las extremidades y huesos de los dedos.
Frecuencia
Los tumores óseos benignos son mucho más comunes que malignas.
Este es el más común de tumor benigno de Osteocondroma condroblastoma y dar condromixoide. Conecte el contrario, con menos frecuencia.
Causas
Las razones para el desarrollo de los tumores óseos benignos son todavía en gran parte desconocida.
En casos raros, las bases genéticas juegan un papel importante.
Algunos tipos de cáncer son hereditarios rara vez se muestra, se incluyen osteocondromas.
Si los factores físicos, químicos o bioquímicos implicados en el desarrollo y aún no ha bekannt.Die causas para el desarrollo de los tumores óseos benignos son todavía en gran parte desconocida. En casos raros, las bases genéticas juegan un papel importante.
Algunos tipos de cáncer son hereditarios rara vez se muestra, se incluyen osteocondromas.
Ya sea física, química o bioquímica de factores están involucrados en el desarrollo no se conoce.
Los síntomas
Dependiendo del tipo de tumor, el asiento y diseminarse puede variar, los síntomas de los tumores óseos benignos. Casi todos los pacientes se quejan de dolor en las zonas afectadas.
A menudo, la deformación de las articulaciones o los huesos pueden ser observadas.
Y fracturas óseas causadas por el crecimiento del tumor se producen.
En la infancia puede llevar a problemas de crecimiento.
Los tumores óseos benignos se producen en los lugares preferidos en el cuerpo.
Las costillas son casi nunca cambia tumorales, mientras que por ejemplo, Los osteocondromas son más frecuentes en los puntos cercanos a las articulaciones de las extremidades, especialmente las piernas.
Diagnóstico
El diagnóstico presuntivo fue planteada por la historia (entrevista) con un cuestionario detallado acerca de los síntomas, el curso anterior de la historia de la enfermedad y la familia.
En el examen físico, el dolor y la deformación puede ser evidente.
Para confirmar el diagnóstico, una imagen de rayos X se produce en dos niveles.
Aquí, los cambios óseos típicos se observan.
En los resultados poco claros, por ejemplo sobre la cuestión de si se trata de tumores benignos o malignos, otros estudios como la tomografía computarizada (TC) y resonancia magnética (IRM) se llevan a cabo.
Si no es el diagnóstico sobre la base de la proyección de imagen para dejar en claro, una muestra (biopsia) se recomienda.
El diagnóstico presuntivo fue planteada por la historia (entrevista) con un cuestionario detallado acerca de los síntomas, el curso anterior de la historia de la enfermedad y la familia.
En el examen físico, el dolor y la deformación puede ser evidente.
Para confirmar el diagnóstico, una imagen de rayos X se produce en dos niveles.
Aquí, los cambios óseos típicos se observan.
En los resultados poco claros, por ejemplo sobre la cuestión de si se trata de tumores benignos o malignos, otros estudios como la tomografía computarizada (TC) y resonancia magnética (IRM) se llevan a cabo.
Si no es el diagnóstico sobre la base de la proyección de imagen para dejar en claro, una muestra (biopsia) se recomienda.
Terapia
Dependiendo del tipo de tumor y diseminarse puede variar el tratamiento.
Es, sin embargo, generalmente a partir de la extirpación quirúrgica del tumor.
Con poco tumores sintomáticos bajo la estrecha supervisión también puede ser una de esperar y ser ingeridos.
Fracturas de huesos se puede tratar mediante intervención quirúrgica o conservadora.
Los osteocondromas
El tratamiento quirúrgico se debe hacer si el tumor es sintomático.
Si hay un aumento de tamaño después de el crecimiento, por lo que la operación también se recomienda, como puede ocurrir en casos raros, un desarrollo de un tumor maligno.
Cuidado Posterior
El seguimiento se basa siempre en el tipo de tumor y con el médico a consultar.
Pronóstico
El pronóstico de los tumores óseos benignos generalmente es bueno.
Con el objetivo de la detección precoz de la recurrencia del tumor hasta los exámenes se recomiendan.
Sólo el Exostosenkrankheit tendencia familiar en algunos casos a convertirse en malignos.
Si en estos pacientes después de la finalización de crecimiento se producen un aumento en el tamaño de la exostosis, una nueva investigación / operación se recomienda.
El pronóstico es aún estima que será favorable.
jueves, 30 de septiembre de 2010
Berlín: Andrea Vortkamp & Instituto Max PlancK,
http://www.datoanuncios.org/?a=26795
" exostosis cartilaginosas múltiples "(Inglés herencia múltiple exostosis , HME ) - detrás del extraño nombre significa una enfermedad hereditaria poco frecuente de crecimiento de los huesos.
Aproximadamente 1 de cada 50.000 personas se ve afectada , tumores benignos del hueso (exostosis ) que se producen sobre todo las rodillas , los codos , las costillas o las manos, son el resultado.
Dependiendo del tamaño , ubicación y número de exostosis , los efectos son diferentes, en los casos graves la extirpación quirúrgica inevitable.
Andrea Vortkamp y sus colegas en el Instituto Max -Planck de Genética Molecular de Berlín ocupan de la investigación de los procesos moleculares que controlan la formación (diferenciación) del esqueleto de los mamíferos.
Una molécula clave aquí es "Indian Hedgehog (IHH ), que regula la diferenciación de las células de cartílago del esqueleto en crecimiento y su transformación en células óseas.
Los científicos pudieron demostrar que la señal Ihh es activado por una falta de heparán sulfato , un polisacárido (polisacáridos ), que es una parte sustancial de los tejidos del cartílago de crecimiento.
La unión de heparán sulfato de señalización diferentes factores capaces de vías de señalización como el de influir en la de Ihh .
Para la formación de heparán sulfato, la enzima que necesita el organismo Exostosin 1(EXT 1) . Los investigadores han sabido desde hace algún tiempo que las mutaciones del gen Exostosin1 es una causa de MHE.
Que los ratones transgénicos con un defecto en el gen Exostosin1 (EXT 1) tienen cantidades reducidas de heparán sulfato .
El uso de estos ratones mostraron a Vortkamp y a su personal una cantidad reducida de heparán sulfato en los resultados de una activación del sistema de señalización Ihh .
Esta es probablemente la causa molecular del crecimiento excesivo del hueso y no directa , lo que conduce a la exostosis MHE típico.
" exostosis cartilaginosas múltiples "(Inglés herencia múltiple exostosis , HME ) - detrás del extraño nombre significa una enfermedad hereditaria poco frecuente de crecimiento de los huesos.
Aproximadamente 1 de cada 50.000 personas se ve afectada , tumores benignos del hueso (exostosis ) que se producen sobre todo las rodillas , los codos , las costillas o las manos, son el resultado.
Dependiendo del tamaño , ubicación y número de exostosis , los efectos son diferentes, en los casos graves la extirpación quirúrgica inevitable.
Andrea Vortkamp y sus colegas en el Instituto Max -Planck de Genética Molecular de Berlín ocupan de la investigación de los procesos moleculares que controlan la formación (diferenciación) del esqueleto de los mamíferos.
Una molécula clave aquí es "Indian Hedgehog (IHH ), que regula la diferenciación de las células de cartílago del esqueleto en crecimiento y su transformación en células óseas.
Los científicos pudieron demostrar que la señal Ihh es activado por una falta de heparán sulfato , un polisacárido (polisacáridos ), que es una parte sustancial de los tejidos del cartílago de crecimiento.
La unión de heparán sulfato de señalización diferentes factores capaces de vías de señalización como el de influir en la de Ihh .
Para la formación de heparán sulfato, la enzima que necesita el organismo Exostosin 1(EXT 1) . Los investigadores han sabido desde hace algún tiempo que las mutaciones del gen Exostosin1 es una causa de MHE.
Que los ratones transgénicos con un defecto en el gen Exostosin1 (EXT 1) tienen cantidades reducidas de heparán sulfato .
El uso de estos ratones mostraron a Vortkamp y a su personal una cantidad reducida de heparán sulfato en los resultados de una activación del sistema de señalización Ihh .
Esta es probablemente la causa molecular del crecimiento excesivo del hueso y no directa , lo que conduce a la exostosis MHE típico.
Bibliografia sobre OMC
http://www.portalesmedicos.com/publicaciones/articles/2089/3/Dolor-de-hombro-en-paciente-con-exostosis-multiple.-A-proposito-de-un-caso-clinico
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico
Autor: M. E. Mesa Rivero Publicado: 27/03/2010
Traumatologia , Imagenes de Radiodiagnostico y Radioterapia , Imagenes de Traumatologia , Casos Clinicos de Traumatologia
Bibliografía:
1. Gupta PP, Agarwal D. A middle-aged manwith persisting chest opacity and permanent bony swelling. CMAJ 2006 Nov 15 ; 31 (24): E920-4
2. Manish Chadha, MS; Arun Pal Singh, MS. Secondary chondrosarcoma of the cuboid bone in a patient with multiple exostosis. Can J Surg, 2008.Vol. 51 (1).
3. Judith VMG Bovée. Multiple osteochondromas. Orphanet Journal of Rare Diseases 2008, 3:3
4. Carolyn M. Sofka, MD& Gregory R. Saboeiro, MD& Robert Schneider, MD. Multiple Hereditary Exostoses. HSSJ (2005) 1:49–51.
5. J V M G Bovée, R J B Sakkers, M J A Geirnaerdt, A H M Taminiau, P C W Hogendoorn. Intermediate grade osteosarcoma and chondrosarcoma
6. Arising in an osteochondroma. A case report of a patient with hereditary multiple exostoses.J Clin Pathol 2002;55:226–229
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico
Autor: M. E. Mesa Rivero Publicado: 27/03/2010
Traumatologia , Imagenes de Radiodiagnostico y Radioterapia , Imagenes de Traumatologia , Casos Clinicos de Traumatologia
Bibliografía:
1. Gupta PP, Agarwal D. A middle-aged manwith persisting chest opacity and permanent bony swelling. CMAJ 2006 Nov 15 ; 31 (24): E920-4
2. Manish Chadha, MS; Arun Pal Singh, MS. Secondary chondrosarcoma of the cuboid bone in a patient with multiple exostosis. Can J Surg, 2008.Vol. 51 (1).
3. Judith VMG Bovée. Multiple osteochondromas. Orphanet Journal of Rare Diseases 2008, 3:3
4. Carolyn M. Sofka, MD& Gregory R. Saboeiro, MD& Robert Schneider, MD. Multiple Hereditary Exostoses. HSSJ (2005) 1:49–51.
5. J V M G Bovée, R J B Sakkers, M J A Geirnaerdt, A H M Taminiau, P C W Hogendoorn. Intermediate grade osteosarcoma and chondrosarcoma
6. Arising in an osteochondroma. A case report of a patient with hereditary multiple exostoses.J Clin Pathol 2002;55:226–229
Exostosis multiple y la mutación cromosómica EXT
http://www.portalesmedicos.com/publicaciones/articles/2089/2/Dolor-de-hombro-en-paciente-con-exostosis-multiple.-A-proposito-de-un-caso-clinico
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico .2
Los osteocondromas solitarios son 6 veces más frecuentes que la exóstosis múltiple.
El 62% de los pacientes afectados, presentan historia familiar positiva para la enfermedad (3).
En nuestro caso, el paciente presenta un antecedente familiar, 2 tíos varones por vía materna, existiendo la sospecha de afectación familiar en 3 generaciones anteriores.
La historia familiar sugiere un patrón de herencia autosómico dominante, con una penetrancia de casi el 100%.
Parece que en el desarrollo de la enfermedad se ha implicado la mutación del gen EXT, que actúa como un gen supresor tumoral.
Dicho gen actúa como productor de glicosiltranferasas, relacionadas con la síntesis de un recubrimiento celular de heparan sulfato, afectando así la difusión del morfogen Hedgehog (5).
Se ha identificado una mutación cromosómica en 3 localizaciones distintas:
EXT 1 (cromosoma 8, locus 8q224.1);
EXT 2 (cromosoma 11, en la región pericéntrica);
EXT 3 (cromosoma 19, locus 19 p),
siendo la mutación del gen EXT 1 la más frecuente cuando existe historia familiar de la enfermedad (1).
El proceso de malignización está representado por una inestabilidad cromosómica, previamente demostrada por un alto porcentaje de pérdida de heterocigosidad, y que implica a varios loci en un amplio rango del DNA.
En el caso presentado no hemos realizado estudio genético.
La proporción de individuos que presentan sintomatología clínica incrementa con la edad, siendo bajo al nacer (5%), aumentado con edad (a los 12 años el 96% ha presentado alguna clínica).
En la edad puberal y, coincidiendo con el cierre del cartílago de crecimiento, es cuando los osteocondromas dejan de crecer (1).
Los síntomas principales de la exóstosis múltiple son el dolor, la limitación funcional de alguna articulación, es frecuente el acortamiento del cúbito que ocasiona una incurvación del radio (39-60% de los casos), así como la dismetría de las extremidades (10-50%), y las deformidades angulares en varo ó valgo de la rodilla (8-33%), como de los tobillos (2-54%).
Con frecuencia se asocia talla baja ¿rizomélica? (37-44%).
En menor proporción podemos encontrarnos con otras manifestaciones osteoarticulares como bursitis, compresión de estructuras vasculonerviosas, fracturas del pedículo, y dolores difusos generalizados. (3)
La complicación más importante es la degeneración maligna en condrosarcoma, que se presenta entre un 0.5-5% de los casos, y que suele manifestarse como dolor y aumento de la masa tumoral; así como engrosamiento de la capa cartilaginosa que recubre al tumor en más de 1 cm.. La degeneración hacia otras lesiones malignas como fibrosarcoma, fribrohistiocitoma maligno y osteosarcoma, se presenta con menor frecuencia (5).
Los osteocondromas son raros en el esqueleto axial, habiéndose descrito algunos casos de compresión medular a nivel de C1, C2 y C5.
Si sospechamos que un paciente padece de exóstosis múltiple, debemos realizar:
1- detallado estudio de la historia familiar,
2.-un estudio radiográfico e histológico tumoral y,
3.-cuando sea posible, un estudio genético.
El estudio radiográfico se inicia mediante un mapa óseo, siendo necesario una TAC para poder valorar con exactitud las lesiones y esclarecer el origen de dichas lesiones (2).
Con el TAC podemos realizar un estudio adecuado de la anatomía ósea regional y de las fracturas de las lesiones exofíticas (5).
La RMN con contraste nos permite identificar tanto la morfología del cartílago que recubre al tumor, como de las partes blandas peritumorales, que nos podrá indicar una posible degeneración maligna (5).
En nuestro caso realizamos un mapa óseo al paciente, así como una TAC de la extremidad superior de ambos húmeros debido a los hallazgos previos radiográficos, con la intención de identificar la extensión y crecimiento de los osteocondromas de estas regiones.
Las imágenes de la RNM nos ayudo a descartar la malignización de las lesiones más bizarras.
Debemos realizar un diagnóstico diferencial con la Displasia Hemimélica Epifisaria (enfermedad de Trévor) y la metacondromatosis, enfermedades que no guardan relación con una mutación del gen EXT; así como diferenciarlo de la enfermedad de Mafucci y Ollier, cuyo patrón de presentación es distinto (3).
El manejo de la exóstosis múltiple, así como su tratamiento, es muy variado.
Cuando dichas lesiones causan dolor, compresión de estructuras adyacentes, deformidad estética ó limitación funcional, está indicado el tratamiento quirúrgico, realizando la exéresis de los osteocondromas que causan estas complicaciones (3).
El tratamiento de la enfermedad del antebrazo es controvertido.
Akita et al. en una serie de 23 pacientes, concluyeron que la osteotomía y el alargamiento de los huesos del antebrazo no era muy beneficiosa.
Así, el procedimiento más adecuado, sería la exéresis del tumor, mejorando la movilidad cuando la lesión afecta al extremo distal del cúbito (3).
Hay que advertir al paciente y sus familiares que debe ser revisado de forma periódica y que en el caso que surja algún cambio en la evolución que solicite una consulta médica de forma inmediata.
Tras la pubertad, los osteocondromas no deben de aumentar de tamaño, así una vez alcanzada la madurez esquelética, se recomienda realizar controles anuales, aunque aún no se ha demostrado la eficacia de dicho seguimiento (3).
En el caso de sufrir la malignización de un osteocondroma, está indicada la resección en bloque, incluyendo la pseudocápsula tumoral y márgenes libres de tumor, ya que así podríamos lograr un buen resultado local y a largo plazo.
A nuestro paciente se le realizó exéresis transpedicular del tumor de escápula derecha; y la exéresis del tumor sésil localizado en metáfisis de húmero izquierdo, cuya rotura era la responsable del dolor que presentaba en hombro izquierdo.
Los osteocondromas son tumores benignos y no afectan la esperanza de vida (3).
El riesgo de malignización hacia osteosarcoma es aproximadamente del 1% y, en caso de producirse, el pronóstico dependerá del grado histológico, siendo la supervivencia a los 10 años del 83% para el grado I y del 29% para el grado III (3).
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico .2
Los osteocondromas solitarios son 6 veces más frecuentes que la exóstosis múltiple.
El 62% de los pacientes afectados, presentan historia familiar positiva para la enfermedad (3).
En nuestro caso, el paciente presenta un antecedente familiar, 2 tíos varones por vía materna, existiendo la sospecha de afectación familiar en 3 generaciones anteriores.
La historia familiar sugiere un patrón de herencia autosómico dominante, con una penetrancia de casi el 100%.
Parece que en el desarrollo de la enfermedad se ha implicado la mutación del gen EXT, que actúa como un gen supresor tumoral.
Dicho gen actúa como productor de glicosiltranferasas, relacionadas con la síntesis de un recubrimiento celular de heparan sulfato, afectando así la difusión del morfogen Hedgehog (5).
Se ha identificado una mutación cromosómica en 3 localizaciones distintas:
EXT 1 (cromosoma 8, locus 8q224.1);
EXT 2 (cromosoma 11, en la región pericéntrica);
EXT 3 (cromosoma 19, locus 19 p),
siendo la mutación del gen EXT 1 la más frecuente cuando existe historia familiar de la enfermedad (1).
El proceso de malignización está representado por una inestabilidad cromosómica, previamente demostrada por un alto porcentaje de pérdida de heterocigosidad, y que implica a varios loci en un amplio rango del DNA.
En el caso presentado no hemos realizado estudio genético.
La proporción de individuos que presentan sintomatología clínica incrementa con la edad, siendo bajo al nacer (5%), aumentado con edad (a los 12 años el 96% ha presentado alguna clínica).
En la edad puberal y, coincidiendo con el cierre del cartílago de crecimiento, es cuando los osteocondromas dejan de crecer (1).
Los síntomas principales de la exóstosis múltiple son el dolor, la limitación funcional de alguna articulación, es frecuente el acortamiento del cúbito que ocasiona una incurvación del radio (39-60% de los casos), así como la dismetría de las extremidades (10-50%), y las deformidades angulares en varo ó valgo de la rodilla (8-33%), como de los tobillos (2-54%).
Con frecuencia se asocia talla baja ¿rizomélica? (37-44%).
En menor proporción podemos encontrarnos con otras manifestaciones osteoarticulares como bursitis, compresión de estructuras vasculonerviosas, fracturas del pedículo, y dolores difusos generalizados. (3)
La complicación más importante es la degeneración maligna en condrosarcoma, que se presenta entre un 0.5-5% de los casos, y que suele manifestarse como dolor y aumento de la masa tumoral; así como engrosamiento de la capa cartilaginosa que recubre al tumor en más de 1 cm.. La degeneración hacia otras lesiones malignas como fibrosarcoma, fribrohistiocitoma maligno y osteosarcoma, se presenta con menor frecuencia (5).
Los osteocondromas son raros en el esqueleto axial, habiéndose descrito algunos casos de compresión medular a nivel de C1, C2 y C5.
Si sospechamos que un paciente padece de exóstosis múltiple, debemos realizar:
1- detallado estudio de la historia familiar,
2.-un estudio radiográfico e histológico tumoral y,
3.-cuando sea posible, un estudio genético.
El estudio radiográfico se inicia mediante un mapa óseo, siendo necesario una TAC para poder valorar con exactitud las lesiones y esclarecer el origen de dichas lesiones (2).
Con el TAC podemos realizar un estudio adecuado de la anatomía ósea regional y de las fracturas de las lesiones exofíticas (5).
La RMN con contraste nos permite identificar tanto la morfología del cartílago que recubre al tumor, como de las partes blandas peritumorales, que nos podrá indicar una posible degeneración maligna (5).
En nuestro caso realizamos un mapa óseo al paciente, así como una TAC de la extremidad superior de ambos húmeros debido a los hallazgos previos radiográficos, con la intención de identificar la extensión y crecimiento de los osteocondromas de estas regiones.
Las imágenes de la RNM nos ayudo a descartar la malignización de las lesiones más bizarras.
Debemos realizar un diagnóstico diferencial con la Displasia Hemimélica Epifisaria (enfermedad de Trévor) y la metacondromatosis, enfermedades que no guardan relación con una mutación del gen EXT; así como diferenciarlo de la enfermedad de Mafucci y Ollier, cuyo patrón de presentación es distinto (3).
El manejo de la exóstosis múltiple, así como su tratamiento, es muy variado.
Cuando dichas lesiones causan dolor, compresión de estructuras adyacentes, deformidad estética ó limitación funcional, está indicado el tratamiento quirúrgico, realizando la exéresis de los osteocondromas que causan estas complicaciones (3).
El tratamiento de la enfermedad del antebrazo es controvertido.
Akita et al. en una serie de 23 pacientes, concluyeron que la osteotomía y el alargamiento de los huesos del antebrazo no era muy beneficiosa.
Así, el procedimiento más adecuado, sería la exéresis del tumor, mejorando la movilidad cuando la lesión afecta al extremo distal del cúbito (3).
Hay que advertir al paciente y sus familiares que debe ser revisado de forma periódica y que en el caso que surja algún cambio en la evolución que solicite una consulta médica de forma inmediata.
Tras la pubertad, los osteocondromas no deben de aumentar de tamaño, así una vez alcanzada la madurez esquelética, se recomienda realizar controles anuales, aunque aún no se ha demostrado la eficacia de dicho seguimiento (3).
En el caso de sufrir la malignización de un osteocondroma, está indicada la resección en bloque, incluyendo la pseudocápsula tumoral y márgenes libres de tumor, ya que así podríamos lograr un buen resultado local y a largo plazo.
A nuestro paciente se le realizó exéresis transpedicular del tumor de escápula derecha; y la exéresis del tumor sésil localizado en metáfisis de húmero izquierdo, cuya rotura era la responsable del dolor que presentaba en hombro izquierdo.
Los osteocondromas son tumores benignos y no afectan la esperanza de vida (3).
El riesgo de malignización hacia osteosarcoma es aproximadamente del 1% y, en caso de producirse, el pronóstico dependerá del grado histológico, siendo la supervivencia a los 10 años del 83% para el grado I y del 29% para el grado III (3).
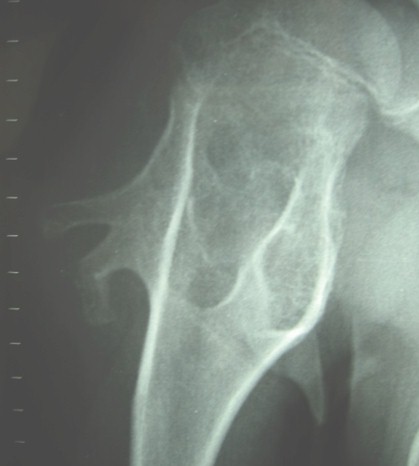{kind=link}
Shoulder pain in a patient with multiple exostoses
http://www.portalesmedicos.com/publicaciones/articles/2089/1/Dolor-de-hombro-en-paciente-con-exostosis-multiple-A-proposito-de-un-caso-clinico.html
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico
Autor: M. E. Mesa Rivero Publicado: 27/03/2010
Traumatologia , Imagenes de Radiodiagnostico y Radioterapia , Imagenes de Traumatologia , Casos Clinicos de Traumatologia
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico.
Mesa Rivero M. E. (MIR), Rodríguez de la Cueva J. M. (FEA), González Herranz J. (FEA), Angulo Gutiérrez J. (FEA).
Servicio de Cirugía Ortopédica y Traumatología. Hospital Universitario de Valme. Sevilla (España).
Dolor de hombro en paciente con exóstosis múltiple. A propósito de 1 caso.
1. Resumen:
Objetivo: La exóstosis hereditaria múltiple es una enfermedad autosómica dominante caracterizada por la aparición de osteocondromas, de predominio metafisario de los huesos largos (1) y que presentan un recubrimiento cartilaginoso benigno.
Los osteocondromas crecen y se desarrollan en la primera década de la vida.
Suelen ser asintomáticos, aunque pueden ocasionar dolor local y compresión de estructuras vasculonerviosas adyacentes o degeneración maligna hacia condrosarcoma, en un 0.5-5% de los casos (2).
Material y método:
Presentamos el caso de un paciente de 12 años de edad, afectado de exóstosis múltiple, que acude a nuestras consultas con dolor en hombro izquierdo.
Se realizó tratamiento mediante exéresis de un pedículo tumoral, desapareciendo la clínica y evolucionando favorablemente.
Conclusión: En el estudio anatomopatológico se diagnóstico de osteocondromas múltiples. Consideramos la importancia del caso debido al potencial de malignización de estas lesiones así como la necesidad de seguimiento genésico de los pacientes.
2. Abstract:
Hereditary multiple exostoses is an autosomal dominant inheritance pattern, characteriszed by benign cartilage-capped bone tumours (osteochondromas) that grow outward from the metaphyses of the long bones.
Osteochondromas develop and increase in size in the first decade of life. The majority are asymptomatic, they may even cause pain and impingement on adjacents nerves and vessels or malignant transformation of osteochondroma towards chondrosarcoma (0.5-5%).
We present a case of a 12 year-old patient with multiple exostoses and left shoulder pain. We resected a humeral tumour. The symptoms disappeared, with a good clinic evolution. Anatomicopathologists carried out a diagnosis of multiple osteochondromas. We consider the relevance of this case because of the malignant degeneration. Patients should be well instructed and followed-up for detection of malignancy.
.
3. Introducción:
La exóstosis hereditaria múltiple constituye una entidad caracterizada por el desarrollo de 20 ó más tumores de localización metafisaria y que, típicamente, presentan un recubrimiento cartilaginoso (1).
Su prevalencia es del 1 entre 50.000 habitantes, siendo más frecuente en los varones (2).
En su origen está implicada la mutación de los genes EXT1 Y EXT2, presentando un patrón de herencia autosómica dominante (3).
Los osteocondromas crecen y se desarrollan en la primera década de la vida, con un número de localizaciones variables entre los individuos de una misma familia.
Se localizan principalmente en la metáfisis de los huesos largos, sobre todo próximos a la rodilla. La mayoría son asintomáticos aunque, en una menor proporción pueden producir dolor, compresión de estructuras adyacentes, deformidad, problemas funcionales y degeneración maligna hacia condrosarcoma, justificando dicha sintomatología la exéresis tumoral (2).
El diagnóstico se realiza mediante la clínica del paciente, apoyado por la pruebas de imagen (4) y, a ser posible, complementándose con la historia familiar y el estudio genético de la misma.
El tratamiento quirúrgico mediante la exéresis tumoral está indicado cuando aparecen complicaciones derivadas por un excesivo crecimiento ó por la malignización.
Los osteocondromas son lesiones benignas, dependiendo así el pronóstico de la degeneración maligna hacia condrosarcoma (5).
El patrón de transmisión es autosómico dominante, con una penetrancia del 100%; así, en caso de aislarse el gen afectado, existe la posibilidad de realizar el diagnóstico prenatal (2).
4. Material y método:
Paciente varón de 12 años de edad, que acude al Servicio de Urgencias de Traumatología al presentar dolor en hombro izquierdo de inicio súbito y sin traumatismo previo. No existen antecedentes personales de interés.
Como antecedentes familiares refiere que 2 hermanos de su madre han sido diagnosticados de exostosis múltiple, sin datos de degeneración maligna en ninguno de ellos.
A la exploración presenta una talla baja para su edad, con extremidades relativamente cortas con respecto al tronco, así como prominencias óseas a nivel de las rodillas, muñecas y hombros. Consideramos como significativo una limitación de la prono-supinación en ambos extremidades superiores.
A la palpación refiere dolor en la zona deltoidea de miembro superior izquierdo y limitación de las rotaciones en ambas articulaciones coxo-femorales.
Realizamos un mapa óseo, hallándose lesiones óseas de predominio metafisario en las tibias, fémures y húmeros, compatibles con el diagnostico de osteocondromatosis.
Destaca la presencia de una fractura del pedículo de un osteocondroma en la metáfisis del húmero izquierdo (figura 1), así como de un ensanchamiento de la metáfisis del húmero derecho debido a la presencia de varios osteocondromas.
Ampliamos el estudio realizando una TAC de la extremidad proximal de ambos húmeros, donde lo más destacado es la fractura del pedículo óseo antes mencionado en el húmero izquierdo, y la presencia de múltiples osteocondromas en la región proximal de húmero y escápula derecha, llamando especialmente la atención la presencia de un osteocondroma con mayor densidad ósea, en la cara interna de la escápula, que separa a esta de la pared torácica (figura 2).
En la RMN no se observan, signos inflamatorios, formación de alguna colección, o otras alteraciones en las partes blandas que nos hagan sospechar malignización del proceso.
Realizamos la exéresis transpedicular de 2 osteocondromas en escápula derecha; así como la extirpación, por vía deltopectoral, de 3 tumores en el extremo proximal del húmero izquierdo.
El material obtenido es remitido como material de biopsia para estudio anatomo-patológico.
5. Resultado:
El resultado del estudio anatomopatológico, reveló la presencia de lesiones de tipo osteocartilaginoso, con centro compuesto por material osteoide y recubrimiento superficial de cartílago. Siendo diagnosticado de osteocondromas.
El paciente fue dado de alta con tratamiento analgésicos habitual y, realizando controles periódicos para el seguimiento de su proceso en régimen ambulatorio y evitar una demora en el diagnostico de una posible degeneración maligna.
6. Discusión:
La exóstosis múltiple es una enfermedad que presenta típicamente tumores (osteocondromas) de predominio metafisario y recubiertos por cartílago, apareciendo sobre todo en los huesos largos (1).
La prevalencia es variable, siendo de 1 cada 100.000 individuos en Europa y de 1/50.000 en el estado de Washington (1).
Son más frecuentes en el varón, existiendo una relación de 1.5: 1, entre hombres y mujeres.
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico
Autor: M. E. Mesa Rivero Publicado: 27/03/2010
Traumatologia , Imagenes de Radiodiagnostico y Radioterapia , Imagenes de Traumatologia , Casos Clinicos de Traumatologia
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico.
Mesa Rivero M. E. (MIR), Rodríguez de la Cueva J. M. (FEA), González Herranz J. (FEA), Angulo Gutiérrez J. (FEA).
Servicio de Cirugía Ortopédica y Traumatología. Hospital Universitario de Valme. Sevilla (España).
Dolor de hombro en paciente con exóstosis múltiple. A propósito de 1 caso.
1. Resumen:
Objetivo: La exóstosis hereditaria múltiple es una enfermedad autosómica dominante caracterizada por la aparición de osteocondromas, de predominio metafisario de los huesos largos (1) y que presentan un recubrimiento cartilaginoso benigno.
Los osteocondromas crecen y se desarrollan en la primera década de la vida.
Suelen ser asintomáticos, aunque pueden ocasionar dolor local y compresión de estructuras vasculonerviosas adyacentes o degeneración maligna hacia condrosarcoma, en un 0.5-5% de los casos (2).
Material y método:
Presentamos el caso de un paciente de 12 años de edad, afectado de exóstosis múltiple, que acude a nuestras consultas con dolor en hombro izquierdo.
Se realizó tratamiento mediante exéresis de un pedículo tumoral, desapareciendo la clínica y evolucionando favorablemente.
Conclusión: En el estudio anatomopatológico se diagnóstico de osteocondromas múltiples. Consideramos la importancia del caso debido al potencial de malignización de estas lesiones así como la necesidad de seguimiento genésico de los pacientes.
2. Abstract:
Hereditary multiple exostoses is an autosomal dominant inheritance pattern, characteriszed by benign cartilage-capped bone tumours (osteochondromas) that grow outward from the metaphyses of the long bones.
Osteochondromas develop and increase in size in the first decade of life. The majority are asymptomatic, they may even cause pain and impingement on adjacents nerves and vessels or malignant transformation of osteochondroma towards chondrosarcoma (0.5-5%).
We present a case of a 12 year-old patient with multiple exostoses and left shoulder pain. We resected a humeral tumour. The symptoms disappeared, with a good clinic evolution. Anatomicopathologists carried out a diagnosis of multiple osteochondromas. We consider the relevance of this case because of the malignant degeneration. Patients should be well instructed and followed-up for detection of malignancy.
.
3. Introducción:
La exóstosis hereditaria múltiple constituye una entidad caracterizada por el desarrollo de 20 ó más tumores de localización metafisaria y que, típicamente, presentan un recubrimiento cartilaginoso (1).
Su prevalencia es del 1 entre 50.000 habitantes, siendo más frecuente en los varones (2).
En su origen está implicada la mutación de los genes EXT1 Y EXT2, presentando un patrón de herencia autosómica dominante (3).
Los osteocondromas crecen y se desarrollan en la primera década de la vida, con un número de localizaciones variables entre los individuos de una misma familia.
Se localizan principalmente en la metáfisis de los huesos largos, sobre todo próximos a la rodilla. La mayoría son asintomáticos aunque, en una menor proporción pueden producir dolor, compresión de estructuras adyacentes, deformidad, problemas funcionales y degeneración maligna hacia condrosarcoma, justificando dicha sintomatología la exéresis tumoral (2).
El diagnóstico se realiza mediante la clínica del paciente, apoyado por la pruebas de imagen (4) y, a ser posible, complementándose con la historia familiar y el estudio genético de la misma.
El tratamiento quirúrgico mediante la exéresis tumoral está indicado cuando aparecen complicaciones derivadas por un excesivo crecimiento ó por la malignización.
Los osteocondromas son lesiones benignas, dependiendo así el pronóstico de la degeneración maligna hacia condrosarcoma (5).
El patrón de transmisión es autosómico dominante, con una penetrancia del 100%; así, en caso de aislarse el gen afectado, existe la posibilidad de realizar el diagnóstico prenatal (2).
4. Material y método:
Paciente varón de 12 años de edad, que acude al Servicio de Urgencias de Traumatología al presentar dolor en hombro izquierdo de inicio súbito y sin traumatismo previo. No existen antecedentes personales de interés.
Como antecedentes familiares refiere que 2 hermanos de su madre han sido diagnosticados de exostosis múltiple, sin datos de degeneración maligna en ninguno de ellos.
A la exploración presenta una talla baja para su edad, con extremidades relativamente cortas con respecto al tronco, así como prominencias óseas a nivel de las rodillas, muñecas y hombros. Consideramos como significativo una limitación de la prono-supinación en ambos extremidades superiores.
A la palpación refiere dolor en la zona deltoidea de miembro superior izquierdo y limitación de las rotaciones en ambas articulaciones coxo-femorales.
Realizamos un mapa óseo, hallándose lesiones óseas de predominio metafisario en las tibias, fémures y húmeros, compatibles con el diagnostico de osteocondromatosis.
Destaca la presencia de una fractura del pedículo de un osteocondroma en la metáfisis del húmero izquierdo (figura 1), así como de un ensanchamiento de la metáfisis del húmero derecho debido a la presencia de varios osteocondromas.
Ampliamos el estudio realizando una TAC de la extremidad proximal de ambos húmeros, donde lo más destacado es la fractura del pedículo óseo antes mencionado en el húmero izquierdo, y la presencia de múltiples osteocondromas en la región proximal de húmero y escápula derecha, llamando especialmente la atención la presencia de un osteocondroma con mayor densidad ósea, en la cara interna de la escápula, que separa a esta de la pared torácica (figura 2).
En la RMN no se observan, signos inflamatorios, formación de alguna colección, o otras alteraciones en las partes blandas que nos hagan sospechar malignización del proceso.
Realizamos la exéresis transpedicular de 2 osteocondromas en escápula derecha; así como la extirpación, por vía deltopectoral, de 3 tumores en el extremo proximal del húmero izquierdo.
El material obtenido es remitido como material de biopsia para estudio anatomo-patológico.
5. Resultado:
El resultado del estudio anatomopatológico, reveló la presencia de lesiones de tipo osteocartilaginoso, con centro compuesto por material osteoide y recubrimiento superficial de cartílago. Siendo diagnosticado de osteocondromas.
El paciente fue dado de alta con tratamiento analgésicos habitual y, realizando controles periódicos para el seguimiento de su proceso en régimen ambulatorio y evitar una demora en el diagnostico de una posible degeneración maligna.
6. Discusión:
La exóstosis múltiple es una enfermedad que presenta típicamente tumores (osteocondromas) de predominio metafisario y recubiertos por cartílago, apareciendo sobre todo en los huesos largos (1).
La prevalencia es variable, siendo de 1 cada 100.000 individuos en Europa y de 1/50.000 en el estado de Washington (1).
Son más frecuentes en el varón, existiendo una relación de 1.5: 1, entre hombres y mujeres.
España: Registro de enfermedades raras (IIER)
Estimada Asociación:
Hace 1 año nos dirigimos a todas las Asociaciones para notificaros la puesta en marcha del Registro de Enfermedades Raras del Instituto de Investigación de Enfermedades Raras (IIER) del Instituto de Salud Carlos III (ISCIII).
El Registro es un instrumento muy útil para la planificación sanitaria y especialmente para la investigación de las Enfermedades Raras y pretende servir de registro específico de cada una de estas enfermedades.
Pasado 1 año en el que hemos comenzado con mucha ilusión y con no pocas dificultades y durante el que hemos ido aprendiendo y mejorando, los resultados empiezan a verse poco a poco. Sin embargo para que el Registro llegue a ser lo que debe ser es imprescindible la colaboración de los pacientes y nosotros no lo podemos lograr sin la inestimable ayuda vuestra, de todas y cada una de las Asociaciones.
En vuestra mano está la posibilidad de comunicar a los pacientes la existencia del Registro y de facilitarles la información necesaria para que puedan solicitar su inscripción en él ó para que puedan ponerse en contacto con nosotros para que les aclaremos cualquier duda o incluso les ayudemos a inscribirse.
Os enviamos un archivo con una pequeña presentación del Registro en la que se explican las características del mismo y como se puede hacer la solicitud para inscribirse a través de la página Web del Registro https://registroraras.isciii.es .
También os enviamos un archivo con la ficha de solicitud y otro archivo con el consentimiento informado por si hay pacientes que desean inscribirse por correo postal.
Os recordamos que podéis poneros en contacto con nosotros a través de este email registro.raras@isciii.es o también directamente con Ángela Almansa en el teléfono 918222050 y en el email aalmansa@isciii.es
Entre todos haremos que el Registro crezca y sea enormemente útil.
Un afectuoso saludo y muchas gracias por vuestra colaboración que no dudamos que nos vais a brindar.
Registro de Enfermedades Raras.
Hace 1 año nos dirigimos a todas las Asociaciones para notificaros la puesta en marcha del Registro de Enfermedades Raras del Instituto de Investigación de Enfermedades Raras (IIER) del Instituto de Salud Carlos III (ISCIII).
El Registro es un instrumento muy útil para la planificación sanitaria y especialmente para la investigación de las Enfermedades Raras y pretende servir de registro específico de cada una de estas enfermedades.
Pasado 1 año en el que hemos comenzado con mucha ilusión y con no pocas dificultades y durante el que hemos ido aprendiendo y mejorando, los resultados empiezan a verse poco a poco. Sin embargo para que el Registro llegue a ser lo que debe ser es imprescindible la colaboración de los pacientes y nosotros no lo podemos lograr sin la inestimable ayuda vuestra, de todas y cada una de las Asociaciones.
En vuestra mano está la posibilidad de comunicar a los pacientes la existencia del Registro y de facilitarles la información necesaria para que puedan solicitar su inscripción en él ó para que puedan ponerse en contacto con nosotros para que les aclaremos cualquier duda o incluso les ayudemos a inscribirse.
Os enviamos un archivo con una pequeña presentación del Registro en la que se explican las características del mismo y como se puede hacer la solicitud para inscribirse a través de la página Web del Registro https://registroraras.isciii.es .
También os enviamos un archivo con la ficha de solicitud y otro archivo con el consentimiento informado por si hay pacientes que desean inscribirse por correo postal.
Os recordamos que podéis poneros en contacto con nosotros a través de este email registro.raras@isciii.es o también directamente con Ángela Almansa en el teléfono 918222050 y en el email aalmansa@isciii.es
Entre todos haremos que el Registro crezca y sea enormemente útil.
Un afectuoso saludo y muchas gracias por vuestra colaboración que no dudamos que nos vais a brindar.
Registro de Enfermedades Raras.
miércoles, 25 de agosto de 2010
Hospital de Jaén: Servicios de análisis genético
http://www.europapress.es/andalucia/sevilla-00357/noticia-hospital-jaen-suma-servicios-analisis-genetico-mutaciones-causan-fribrosis-quistica-20100616184657.html
El Hospital de Jaén suma a sus servicios el análisis genético de las mutaciones que causan la fribrosis quística
JAÉN, 16 Jun. (EUROPA PRESS) -
El Complejo Hospitalario de Jaén ha incorporado en su cartera de servicios el análisis genético de las mutaciones que causan fibrosis quística en pacientes con el objetivo de promover la deteccion precoz y la instauración de un tratamiento temprano integral, que pueda mejorar tanto la supervivencia como la calidad de vida.
El test, según informó hoy el Complejo en un comunicado, se realiza en una muestra de sangre periférica en niños y bebes con problemas respiratorios asociados con manifestaciones digestivas o en aquellas personas con antecedentes familiares de esta enfermedad.
En la actualidad, y desde finales del año 2009, la Unidad de Gestión Clínica de Laboratorio del hospital jiennense está analizando las 36 mutaciones de fibrosis quística más frecuentes descrita en nuestra población.
La detección precoz de esta enfermedad mediante el estudio genético es importante, ya que permite aplicar medidas que contribuyen a reducir sus consecuencias.
Además, el estudio genético de las mutaciones causantes de fibrosis quística hace posible la realización de consejo genético.
La Fibrosis Quística es una enfermedad genética que afecta, en especial medida, al aparato respiratorio y digestivo, causando discapacidad progresiva y muerte prematura, por la acumulación de un moco espeso y pegajoso en los pulmones y tubo digestivo.
Es uno de los tipos de enfermedad pulmonar crónica más común en niños y adultos jóvenes.
Se manifiesta en edades muy tempranas y se considera una de las enfermedades hereditarias más comunes.
1 de cada 25 personas es portadora del gen defectuoso de la fibrosis quística, pudiéndola trasmitir a sus hijos si coincide con otro progenitor también portador de la misma patología.
En España afecta a 3.000 personas y a cerca de medio millar en Andalucía.
La esperanza de vida media de los pacientes de esta enfermedad es de aproximadamente 35 a 40 años.
Un diagnóstico precoz y un tratamiento adecuado aminoran los efectos de la patología y mejoran la calidad de vida.
Los afectados pueden ser diagnosticados mediante pruebas genéticas prenatales, también por 'screening' neonatal o durante la infancia temprana.
La Consejería de Salud cuenta entre las medidas sobre este tipo de dolencias realizar estudios genéticos a todos los recién nacidos como prueba de screening, tal y como se contempla en el Plan de Atención a Personas Afectadas por enfermedades raras (2008-2012).
BIOLOGÍA MOLECULAR
El Complejo Hospitalario de Jaén cuenta con una nueva Unidad de Biología Molecular que ha realizado en torno a las 5.500 determinaciones durante su 1º año de funcionamiento, una actividad que ha permitido la realización de diferentes técnicas que ayudan al diagnóstico, la prevención, elección de tratamiento y la evaluación de riesgo de diferentes enfermedades.
Entre los trabajos que se están realizando actualmente se encuentran estudios genéticos de hemocromatosis hereditaria, trombofilias, marcador genético de celiaquía, análisis antígenos de histocompatibilidad y análisis de traslocaciones cromosómicas, relacionadas con enfermedades oncohematológicas, o fibrosis quística.
La Unidad está dotada con los más modernos equipos y cuenta con personal especializado, en concreto con las profesionales Esther Ocaña, Rosario Aguilar y la responsable del Servicio de Análisis Clínicos, Manuela Gassó.
"La aplicación de estas técnicas de Biología Molecular permitirá la aplicación de tratamientos personalizados en procesos tumorales, que hace algunos años era imposible de imaginar, y la realización de nuevas pruebas a los recién nacidos para la detección precoz de enfermedades raras, como la fibrosis quística, siendo esta una de las líneas prioritarias de la Consejería de Salud", destacó Gassó, que incidió en que, además, la unidad propiciará la solicitud de proyectos que requieran métodos de Biología Molecular.
Hasta el momento, la realización de algunas pruebas genéticas especiales se estaban realizando en laboratorios externos al centro.
Con la implantación de esta Unidad, se ha conseguido realizar estas pruebas en el Complejo Hospitalario de Jaén, permitiendo ofrecer este servicio al resto de hospitales de la provincia.
El Hospital de Jaén suma a sus servicios el análisis genético de las mutaciones que causan la fribrosis quística
JAÉN, 16 Jun. (EUROPA PRESS) -
El Complejo Hospitalario de Jaén ha incorporado en su cartera de servicios el análisis genético de las mutaciones que causan fibrosis quística en pacientes con el objetivo de promover la deteccion precoz y la instauración de un tratamiento temprano integral, que pueda mejorar tanto la supervivencia como la calidad de vida.
El test, según informó hoy el Complejo en un comunicado, se realiza en una muestra de sangre periférica en niños y bebes con problemas respiratorios asociados con manifestaciones digestivas o en aquellas personas con antecedentes familiares de esta enfermedad.
En la actualidad, y desde finales del año 2009, la Unidad de Gestión Clínica de Laboratorio del hospital jiennense está analizando las 36 mutaciones de fibrosis quística más frecuentes descrita en nuestra población.
La detección precoz de esta enfermedad mediante el estudio genético es importante, ya que permite aplicar medidas que contribuyen a reducir sus consecuencias.
Además, el estudio genético de las mutaciones causantes de fibrosis quística hace posible la realización de consejo genético.
La Fibrosis Quística es una enfermedad genética que afecta, en especial medida, al aparato respiratorio y digestivo, causando discapacidad progresiva y muerte prematura, por la acumulación de un moco espeso y pegajoso en los pulmones y tubo digestivo.
Es uno de los tipos de enfermedad pulmonar crónica más común en niños y adultos jóvenes.
Se manifiesta en edades muy tempranas y se considera una de las enfermedades hereditarias más comunes.
1 de cada 25 personas es portadora del gen defectuoso de la fibrosis quística, pudiéndola trasmitir a sus hijos si coincide con otro progenitor también portador de la misma patología.
En España afecta a 3.000 personas y a cerca de medio millar en Andalucía.
La esperanza de vida media de los pacientes de esta enfermedad es de aproximadamente 35 a 40 años.
Un diagnóstico precoz y un tratamiento adecuado aminoran los efectos de la patología y mejoran la calidad de vida.
Los afectados pueden ser diagnosticados mediante pruebas genéticas prenatales, también por 'screening' neonatal o durante la infancia temprana.
La Consejería de Salud cuenta entre las medidas sobre este tipo de dolencias realizar estudios genéticos a todos los recién nacidos como prueba de screening, tal y como se contempla en el Plan de Atención a Personas Afectadas por enfermedades raras (2008-2012).
BIOLOGÍA MOLECULAR
El Complejo Hospitalario de Jaén cuenta con una nueva Unidad de Biología Molecular que ha realizado en torno a las 5.500 determinaciones durante su 1º año de funcionamiento, una actividad que ha permitido la realización de diferentes técnicas que ayudan al diagnóstico, la prevención, elección de tratamiento y la evaluación de riesgo de diferentes enfermedades.
Entre los trabajos que se están realizando actualmente se encuentran estudios genéticos de hemocromatosis hereditaria, trombofilias, marcador genético de celiaquía, análisis antígenos de histocompatibilidad y análisis de traslocaciones cromosómicas, relacionadas con enfermedades oncohematológicas, o fibrosis quística.
La Unidad está dotada con los más modernos equipos y cuenta con personal especializado, en concreto con las profesionales Esther Ocaña, Rosario Aguilar y la responsable del Servicio de Análisis Clínicos, Manuela Gassó.
"La aplicación de estas técnicas de Biología Molecular permitirá la aplicación de tratamientos personalizados en procesos tumorales, que hace algunos años era imposible de imaginar, y la realización de nuevas pruebas a los recién nacidos para la detección precoz de enfermedades raras, como la fibrosis quística, siendo esta una de las líneas prioritarias de la Consejería de Salud", destacó Gassó, que incidió en que, además, la unidad propiciará la solicitud de proyectos que requieran métodos de Biología Molecular.
Hasta el momento, la realización de algunas pruebas genéticas especiales se estaban realizando en laboratorios externos al centro.
Con la implantación de esta Unidad, se ha conseguido realizar estas pruebas en el Complejo Hospitalario de Jaén, permitiendo ofrecer este servicio al resto de hospitales de la provincia.
El Clinic y Sant Joan de Déu crean una unidad de adultos con enfermedades raras
http://www.europapress.es/sociedad/salud/noticia-cataluna-clinic-sant-joan-deu-crean-unidad-adultos-enfermedades-raras-20100608124604.html
El Clínic y Sant Joan de Déu crean una unidad de adultos con enfermedades raras
Directorio:Hospital Universitario,enfermedades raras,Investigación Biomédica,
Hospital Clínic
BARCELONA, 8 Jun. (EUROPA PRESS) -
El Hospital Clínic de Barcelona y el Hospital de Sant Joan de Déu en Esplugues de Llobregat (Barcelona) podrán pondrán en marcha una unidad para el diagnóstico y seguimiento de adultos afectados por enfermedades raras, en el marco del Centro de Investigación Biomédica en Red de Enfermedades Raras (Ciberer).
En un comunicado, el Clínic explica que cada vez se diagnostican más casos de enfermedades metabólicas hereditarias y otras patologías minoritarias, al tiempo que los afectados llegan a la edad adulta por la mejora en los tratamientos.
Ello exige equipos multidisciplinares que puedan hacer frente a las complicaciones de tipo neurológico, obstétrico, nutricional o dietético.
El Clínic acoge este martes una jornada sobre Enfermedades Metabólicas Hereditarias en Adultos para abordar los retos en diagnóstico, terapia y seguimiento de los afectados.
Participarán diversos grupos asistenciales implicados en la puesta en marcha de la unidad, así como representantes del Hospital Pitie-Salpêtrière y la Universidad Pierre et Marie Curie de París, uno de los pocos referentes mundiales en la materia.
El Clínic y Sant Joan de Déu crean una unidad de adultos con enfermedades raras
Directorio:Hospital Universitario,enfermedades raras,Investigación Biomédica,
Hospital Clínic
BARCELONA, 8 Jun. (EUROPA PRESS) -
El Hospital Clínic de Barcelona y el Hospital de Sant Joan de Déu en Esplugues de Llobregat (Barcelona) podrán pondrán en marcha una unidad para el diagnóstico y seguimiento de adultos afectados por enfermedades raras, en el marco del Centro de Investigación Biomédica en Red de Enfermedades Raras (Ciberer).
En un comunicado, el Clínic explica que cada vez se diagnostican más casos de enfermedades metabólicas hereditarias y otras patologías minoritarias, al tiempo que los afectados llegan a la edad adulta por la mejora en los tratamientos.
Ello exige equipos multidisciplinares que puedan hacer frente a las complicaciones de tipo neurológico, obstétrico, nutricional o dietético.
El Clínic acoge este martes una jornada sobre Enfermedades Metabólicas Hereditarias en Adultos para abordar los retos en diagnóstico, terapia y seguimiento de los afectados.
Participarán diversos grupos asistenciales implicados en la puesta en marcha de la unidad, así como representantes del Hospital Pitie-Salpêtrière y la Universidad Pierre et Marie Curie de París, uno de los pocos referentes mundiales en la materia.
SEMFYC: Protocolo On Line para pacientes con Enfermedades Raras
http://www.imserso.es/creer_01/documentacion/boletindigitalcreer/ano_2010/julioagosto/monograficomes/index.htm
Protocolo online para la atención de los pacientes con Enfermedades Raras
La semFYC ha elaborado un protocolo online de atención a pacientes con enfermedades raras en la consulta de atención primaria, en colaboración con el Instituto de Investigación de Enfermedades Raras (IIER), perteneciente al Instituto de Salud Carlos III (ISCIII), la Federación Española de Enfermedades Raras (Feder) y el Centro de Referencia Estatal de Atención a Personas con Enfermedades Raras y sus Familias de Burgos (Creer).
CADA MÉDICO DE FAMILIA ATIENDE UNA MEDIA DE 15 PACIENTES AL AÑO CON UNA ENFERMEDAD RARA
3 millones de españoles están afectados por una de estas patologías, que se caracterizan por:
1.- su cronicidad,
2.- baja prevalencia,
3.- elevada morbilidad y mortalidad precoz así como,
4.- en muchos casos, necesidades especiales.
Objetivos:
a.- Mejorar el diagnóstico y la información,
b.- así como la coordinación entre los diferentes especialistas y
c.- facilitar datos epidemiológicos que justifiquen la necesidad de investigar más en estas enfermedades son algunos de los objetivos de este protocolo
El médico de familia debe informar y asesorar al paciente, así como animarle a que entre a formar parte del Registro Epidemiológico del Programa de Investigación en Enfermedades Raras (REpIER), dirigido desde el Instituto de Investigación de Enfermedades Raras.
Madrid, X de junio de 2010 -
Entre 10 y 15 pacientes que pasan al año por la consulta del médico de familia están afectados por una Enfermedad Rara (ER).
Un conjunto de patologías crónicas muy diversas que se caracterizan por su baja prevalencia (menos de 5 casos por cada 10.000 habitantes) elevada morbilidad y mortalidad precoz, lo que determina en muchos casos unas necesidades especiales.
En este contexto, la Sociedad Española de Medicina de Familia y Comunitaria (semFYC), que representa a más de 20.000 profesionales de Atención Primaria de toda España, ha elaborado un protocolo onlinepara la atención de los pacientes con ER en la consulta de Atención Primaria, contando con la colaboración del:
a.- Instituto de Investigación de Enfermedades Raras (IIER), dependiente del Instituto de Salud Carlos III,
b.- de la Federación Española de Enfermedades Raras (Feder) y
c.- del Centro de Referencia Estatal de Atención a Personas con Enfermedades Raras y sus Familias de Burgos (Creer).
Como señala el coordinador del Grupo de Genética Clínica y Enfermedades Raras de semFYC, el Dr. Miguel García Ribes, :
“los objetivos del protocolo son mejorar el diagnóstico y la información que existe en torno a estas patologías, así como la coordinación entre los diferentes especialistas y facilitar datos epidemiológicos que justifiquen la puesta en marcha de acciones en el ámbito de la planificación sanitaria y fomenten la investigación de las ER.
Su baja prevalencia ha condicionado a que existan pocos estudios y los tratamientos en la mayoría de los casos son inexistentes.
Aunque es todavía pronto para asegurar que estas personas tienen cubiertas sus necesidades específicas socio-sanitarias, las autoridades tanto sanitarias como del ámbito social ya empiezan a tomar conciencia de las dimensiones de este problema, que en nuestro país afecta a cerca de tres millones de personas”.
Falta de coordinación entre profesionales
Una de las principales demandas de estos pacientes es la falta de coordinación entre los profesionales.
En este sentido, el protocolo DICE-APER busca mejorar la colaboración asistencial, estableciendo los lazos oportunos con el servicio médico especialista al que ha sido derivado cada paciente.
“En este protocolo”, explica el Doctor García Ribes, “se incluye el envío de una carta, que el médico de familia puede hacer llegar al especialista que lleva cada caso vía correo electrónico o correo postal, en la cual se informará al médico especialista acerca del interés y disposición para que exista una coordinada estrecha entre la atención especializada la correspondiente atención en la consulta de primaria y facilitar sus datos de contacto para poder mantener un intercambio fluido de información sobre la evolución clínica de esa persona”.
Información y diagnóstico
El médico de familia debe confirmar que el diagnóstico que trae el paciente es el de una ER y siempre debe quedar reflejado en su historia clínica.
Asimismo, este profesional juega un papel clave en la educación y asesoría del paciente con una ER.
“Es importante que expliquemos a nuestros pacientes que existe un registro de pacientes de ER y del interés que pertenecer al mismo tiene para la investigación de su enfermedad, por lo que se le invita a formar parte del mismo. El crecimiento de esta base de datos es clave para conocer las dimensiones del problema. Asimismo, el biobanco de ER es una pieza fundamental en la promoción de la investigación y en los análisis que ayuden al diagnóstico y la búsqueda de biomarcadores pronósticos.
Sin embrago, la baja prevalencia de estas patologías dificulta conseguir muestras biológicas que sirvan para llevar a cabo investigaciones de mayos calado científico.
Es imprescindible mostrar al paciente de la importancia de esta donación de una pequeña muestra de sangre, que se haría coincidir con una de sus extracciones analíticas de rutina”.
En opinión del Dr. García Ribes, este protocolo no pretende crear más sobrecarga de trabajo, sino que lo que persigue es ordenar de forma lógica aquellas tareas que podrían ser asumidas por el médico de familia en la atención a estos pacientes.
“De este modo se cubrirían objetivos básicos que facilitarían el control y la gestión de las ER, a la vez que convertiríamos esta faceta de la AP en un modelo de atención exportable a Europa”, concluye este experto.
• Para más información, Gabinete de Prensa semFYC: 91.787.03.00
Protocolo online para la atención de los pacientes con Enfermedades Raras
La semFYC ha elaborado un protocolo online de atención a pacientes con enfermedades raras en la consulta de atención primaria, en colaboración con el Instituto de Investigación de Enfermedades Raras (IIER), perteneciente al Instituto de Salud Carlos III (ISCIII), la Federación Española de Enfermedades Raras (Feder) y el Centro de Referencia Estatal de Atención a Personas con Enfermedades Raras y sus Familias de Burgos (Creer).
CADA MÉDICO DE FAMILIA ATIENDE UNA MEDIA DE 15 PACIENTES AL AÑO CON UNA ENFERMEDAD RARA
3 millones de españoles están afectados por una de estas patologías, que se caracterizan por:
1.- su cronicidad,
2.- baja prevalencia,
3.- elevada morbilidad y mortalidad precoz así como,
4.- en muchos casos, necesidades especiales.
Objetivos:
a.- Mejorar el diagnóstico y la información,
b.- así como la coordinación entre los diferentes especialistas y
c.- facilitar datos epidemiológicos que justifiquen la necesidad de investigar más en estas enfermedades son algunos de los objetivos de este protocolo
El médico de familia debe informar y asesorar al paciente, así como animarle a que entre a formar parte del Registro Epidemiológico del Programa de Investigación en Enfermedades Raras (REpIER), dirigido desde el Instituto de Investigación de Enfermedades Raras.
Madrid, X de junio de 2010 -
Entre 10 y 15 pacientes que pasan al año por la consulta del médico de familia están afectados por una Enfermedad Rara (ER).
Un conjunto de patologías crónicas muy diversas que se caracterizan por su baja prevalencia (menos de 5 casos por cada 10.000 habitantes) elevada morbilidad y mortalidad precoz, lo que determina en muchos casos unas necesidades especiales.
En este contexto, la Sociedad Española de Medicina de Familia y Comunitaria (semFYC), que representa a más de 20.000 profesionales de Atención Primaria de toda España, ha elaborado un protocolo onlinepara la atención de los pacientes con ER en la consulta de Atención Primaria, contando con la colaboración del:
a.- Instituto de Investigación de Enfermedades Raras (IIER), dependiente del Instituto de Salud Carlos III,
b.- de la Federación Española de Enfermedades Raras (Feder) y
c.- del Centro de Referencia Estatal de Atención a Personas con Enfermedades Raras y sus Familias de Burgos (Creer).
Como señala el coordinador del Grupo de Genética Clínica y Enfermedades Raras de semFYC, el Dr. Miguel García Ribes, :
“los objetivos del protocolo son mejorar el diagnóstico y la información que existe en torno a estas patologías, así como la coordinación entre los diferentes especialistas y facilitar datos epidemiológicos que justifiquen la puesta en marcha de acciones en el ámbito de la planificación sanitaria y fomenten la investigación de las ER.
Su baja prevalencia ha condicionado a que existan pocos estudios y los tratamientos en la mayoría de los casos son inexistentes.
Aunque es todavía pronto para asegurar que estas personas tienen cubiertas sus necesidades específicas socio-sanitarias, las autoridades tanto sanitarias como del ámbito social ya empiezan a tomar conciencia de las dimensiones de este problema, que en nuestro país afecta a cerca de tres millones de personas”.
Falta de coordinación entre profesionales
Una de las principales demandas de estos pacientes es la falta de coordinación entre los profesionales.
En este sentido, el protocolo DICE-APER busca mejorar la colaboración asistencial, estableciendo los lazos oportunos con el servicio médico especialista al que ha sido derivado cada paciente.
“En este protocolo”, explica el Doctor García Ribes, “se incluye el envío de una carta, que el médico de familia puede hacer llegar al especialista que lleva cada caso vía correo electrónico o correo postal, en la cual se informará al médico especialista acerca del interés y disposición para que exista una coordinada estrecha entre la atención especializada la correspondiente atención en la consulta de primaria y facilitar sus datos de contacto para poder mantener un intercambio fluido de información sobre la evolución clínica de esa persona”.
Información y diagnóstico
El médico de familia debe confirmar que el diagnóstico que trae el paciente es el de una ER y siempre debe quedar reflejado en su historia clínica.
Asimismo, este profesional juega un papel clave en la educación y asesoría del paciente con una ER.
“Es importante que expliquemos a nuestros pacientes que existe un registro de pacientes de ER y del interés que pertenecer al mismo tiene para la investigación de su enfermedad, por lo que se le invita a formar parte del mismo. El crecimiento de esta base de datos es clave para conocer las dimensiones del problema. Asimismo, el biobanco de ER es una pieza fundamental en la promoción de la investigación y en los análisis que ayuden al diagnóstico y la búsqueda de biomarcadores pronósticos.
Sin embrago, la baja prevalencia de estas patologías dificulta conseguir muestras biológicas que sirvan para llevar a cabo investigaciones de mayos calado científico.
Es imprescindible mostrar al paciente de la importancia de esta donación de una pequeña muestra de sangre, que se haría coincidir con una de sus extracciones analíticas de rutina”.
En opinión del Dr. García Ribes, este protocolo no pretende crear más sobrecarga de trabajo, sino que lo que persigue es ordenar de forma lógica aquellas tareas que podrían ser asumidas por el médico de familia en la atención a estos pacientes.
“De este modo se cubrirían objetivos básicos que facilitarían el control y la gestión de las ER, a la vez que convertiríamos esta faceta de la AP en un modelo de atención exportable a Europa”, concluye este experto.
• Para más información, Gabinete de Prensa semFYC: 91.787.03.00
jueves, 19 de agosto de 2010
Exostosis tibial: Osteocondroma solitario
http://www.pap.es/FrontOffice/PAP/front/Articulos/Articulo/_IXus5l_LjPoo2J2KDAbNm7islArjMMmA
Exostosis tibial: osteocondroma
Autores:
Parada López R a, Montano Navarro E b, Lafraya Puente AL c, Rodríguez Ortega M d
a Pediatra. CS Pinto y Getafe. Servicio Madrileño de Salud, Área 10. Madrid. España.
b Pediatra. CS Pinto y Getafe. Servicio Madrileño de Salud, Área 10. Madrid. España.
c Pediatra. CS Pinto y Getafe. Servicio Madrileño de Salud, Área 10. Madrid. España.
d Cirugía General y Aparato Digestivo. Hospital Infanta Cristina. Parla, Madrid. España.
Correspondencia: R Parada. Correo electrónico: rebecaparada@hotmail.com
Referencia para citar este artículo:
Parada López R, Montano Navarro E, Lafraya Puente AL, Rodríguez Ortega M.
Exostosis tibial: osteocondroma. Rev Pediatr Aten Primaria. 2010;12:255-261.
Resumen:
El osteocondroma o exostosis cartilaginosa es el tumor óseo más frecuente, representando el 10-15% de la totalidad. Alrededor del 3% de la población lo padece.
Es un tumor propio de individuos jóvenes con ligero predominio en varones.
Puede ser solitario o múltiple, formando parte del síndrome de exostosis múltiple hereditaria.
Suele ser un hallazgo accidental, normalmente asintomático, cuya localización más frecuente es la rodilla, aunque puede aparecer en otras localizaciones.
El diagnóstico se realiza por imagen radiográfica, que suele ser patognomónica, y el tratamiento definitivo consiste en la extirpación quirúrgica.
Introducción
Los osteocondromas son lesiones benignas formadoras de cartílago.
Suponen entre el 20-50% de los tumores benignos y entre el 10-20% de todos los tumores primarios del hueso; alrededor del 3% de la población lo padece1-3.
Son de origen cartilaginoso y se conforman tras la separación de un fragmento desde el cartílago epifisario.
Pueden ser solitarios o múltiples, y pueden aparecer espontáneamente o tras un traumatismo. Han sido descritos en prácticamente todos los huesos del esqueleto, pero tienen predilección por la metáfisis de huesos largos.
En este artículo planteamos el caso de un paciente de 13 años de edad que acude a nuestra consulta por presentar una excrecencia a nivel tibial secundaria a este tumor de origen óseo confirmado mediante radiodiagnóstico.
Caso clínico
Varón de 13 años, sin antecedentes personales de interés, que acude a la consulta del centro de salud porque desde hace unos meses ha notado una lesión en la pierna izquierda que no ocasiona ninguna sintomatología y no ha variado de tamaño.
Refiere un posible traumatismo sobre dicha zona 2 años antes tras una caída con patines.
Presenta un buen estado general y se encuentra asintomático.
Se palpa prominencia de aproximadamente 1,5-2 cm de consistencia ósea alrededor de 3-4 cm por encima del maléolo interno izquierdo.
No se objetiva inflamación local, hematoma ni dolor a la palpación.
Tampoco existen alteraciones neurovasculares distales ni impotencia funcional.
Se solicita una radiografía simple de la pierna afecta, anteroposterior y lateral, donde se visualiza una lesión de carácter óseo.
El informe radiológico da el diagnóstico:
“La radiografía anteroposterior y lateral del tobillo muestra un relieve óseo bien delimitado, a expensas de la cortical, sin aparente afectación de partes blandas, en cara medial de tercio distal de la tibia izquierda compatible con osteocondroma”.
Dado que se trata de una masa asintomática y se ha mantenido estable conservando su tamaño, se decide seguir la evolución en la consulta y ante posibles cambios realizar prueba de imagen y/o citarlo en consultas de traumatología para revisión.
Discusión
Nos encontramos ante un caso de osteocondroma, exostosis ósea recubierta de cartílago que se ubica en la superficie externa del hueso.
Es un tumor propio de individuos muy jóvenes; de hecho, el 70% de las lesiones osteocondrales se encuentra en las 2 primeras décadas de la vida2,4.
Tiene un ligero predominio en varones, con una proporción de 3/12.
Los osteocondromas se forman tras la separación de un fragmento desde el cartílago epifisario, que se hernia a través del periostio que envuelve el platillo de crecimiento.
El desarrollo posterior de este fragmento cartilaginoso y su osificación endocondral dará lugar a la exostosis recubierta de cartílago que se proyecta hacia la superficie ósea.
El cartílago extruido sigue el proceso normal de osificación, tiene un espesor menor de 1 cm y es histológicamente normal5.
Los osteocondromas pueden ser solitarios o múltiples1,4, siendo estos últimos constituyentes de la exostosis hereditaria múltiple1,3,6.
En nuestro paciente sólo se encontró una lesión y no había habido ninguna otra persona en su familia con un cuadro similar.
Algunos adolescentes refieren un antecedente de traumatismo o ejercicio vigoroso previo a la aparición del mismo9.
La exostosis puede desarrollarse en cualquier hueso del esqueleto, mostrando preferencia por las zonas metafisarias vecinas a los cartílagos más fértiles de los huesos largos: distal del fémur, proximal de la tibia, distal del radio, etc. La ubicación más frecuente es la rodilla2,4-6.
En ocasiones se desarrollan en huesos de la pelvis, escápula y costillas, tratándose de masas sésiles de pedículo corto. Es raro que afecten a los huesos cortos de manos y pies.
La lesión que comienza precozmente causa trastornos de crecimiento, con acortamiento y deformidad de la extremidad afectada.
Sin embargo, habitualmente es asintomática y se diagnostica como un hallazgo casual1, como el caso que nos ocupa.
Las deformidades esqueléticas y estéticas causadas por los osteocondromas subyacentes son la forma característica de presentación.
Igualmente, pueden producir compresión extrínseca sobre las estructuras óseas, articulares, nerviosas, musculares y ligamentosas vecinas.
También pueden producir compresión de vísceras, como ocurre en los derrames pleurales causados por los osteocondromas costales.
En las zonas de fricción ósea se suelen desarrollar bolsas sinoviales.
Las fracturas son infrecuentes y se producen principalmente en la región de la rodilla2,4,5.
Finalmente, lo más importante a tener en cuenta es que si un osteocondroma duele, además hay que descartar su posible transformación maligna (condrosarcoma)2,3,7.
El riesgo de transformación sarcomatosa en la exostosis solitaria es aproximadamente del 1%, pero en la forma múltiple hereditaria el riesgo se acerca al 10% debido a las numerosas lesiones3. Los datos que nos inducen a pensar que existe una transformación maligna son:
1) la lesión se hace sintomática de repente (duele) o empieza a crecer rápidamente;
2) la cubierta cartilaginosa es más gruesa de 1 cm en un adulto (en el niño puede ser de 2-3 cm de espesor) o con un diámetro máximo mayor de 5 cm;
3) el aumento súbito o marcado en la captación en la gammagrafía ósea en un adulto (en la madurez esquelética lo normal es que esté latente); y
4) la confirmación por la TC (tomografía computadorizada) o RM (resonancia magnética) de una masa de tejidos blandos o desplazamiento de un paquete neurovascular mayor4.
Normalmente los grandes osteocondromas suelen producir desplazamiento de los vasos vecinos. Las complicaciones vasculares de los osteocondromas incluyen: pseudoaneurismas, oclusiones arteriales y venosas y fístulas arteriovenosas.
El 90% de las complicaciones vasculares afectan directamente a las arterias, siendo los pseudoaneurismas la patología más frecuente (63,9%)3-5.
En cuanto al diagnóstico, la prueba de imagen más usada es la radiografía simple.
La apariencia radiográfica de una exostosis es la de una lesión aplanada, sésil o pediculada3-4 (imagen radiológica patognomónica).
Actualmente, la ecografía es el método más accesible en nuestro medio.
Es útil en la valoración del casquete cartilaginoso y además tiene la ventaja de que en los casos de existencia de pseudoaneurisma, muestra con facilidad la presencia de una masa compleja con flujo en su interior y dependiente de una gran arteria.
Otros métodos de imagen son:
la TC (define estructuras óseas anómalas y su relación con complicaciones vasculares mediante la inyección de contraste endovenoso y es de gran utilidad en el estudio del osteocondroma que se origina en hombro, pelvis, columna vertebral y escápula) y la RM, prueba de elección ante la sospecha de malignización. Permite detectar la continuidad entre el hueso medular y cortical del tumor y el hueso afecto. Además el casquete cartilaginoso es hiperintenso en secuencia T21-2.
Cuando en la consulta de Atención Primaria nos encontramos un caso como éste, debemos llevar a cabo un diagnóstico diferencial, aunque su presentación clínica no suele presentar problemas para el diagnóstico. Debemos diferenciarlo principalmente de la exostosis cartilaginosa múltiple y el osteosarcoma parostal.
La exostosis cartilaginosa múltiple es una enfermedad congénita caracterizada por tumores óseos que se distribuyen por casi todo el esqueleto, descrita ya en 1891 por Bessel-Hagen.
Tiene una herencia autosómica dominante.
Se han identificado tres loci en relación con la enfermedad:
EXT1, en el cromosoma 8q23-q24;
EXT2 en el 11p11-p12 y
EXT3 en el brazo corto del cromosoma 19.
Alrededor de 2/3 de los pacientes tienen antecedentes familiares de la enfermedad.
Esta forma múltiple tiene una prevalencia estimada entre el 1/1.000 y el 1/50.000, dependiendo de la zona geográfica que se estudie7.
Se debe buscar esta entidad en pacientes con estatura corta, achatamiento de radio y deformidad angular de los miembros inferiores, signos que no están presentes en nuestro paciente.
El osteosarcoma parostal puede presentarse como una exostosis sintomática que aumenta de tamaño en los adultos.
Está compuesto de tejidos fibro-óseos densos que no están presentes en el osteocondroma.
Con respecto al tratamiento del osteocondroma, existen dos opciones;
a.- por un lado el conservador, en aquellos casos que resultan asintomáticos y se mantienen estables y,
b.-por otro, el quirúrgico, indicado en las formas sintomáticas de la infancia y el crecimiento continuado del osteocondroma después de la madurez del esqueleto (sospecha de malignización)4.
El tratamiento definitivo consiste en una resección simple o ampliada.
Si es pediculado se hace la resección simple hasta la base.
Cuando es sésil se hace una resección ampliada sacando una zona de cortical y completando con curetaje 6, extirpando siempre la exostosis, su cobertura cartilaginosa y el periostio, ya que los restos de éste y del pericondrio pueden dar lugar a una recidiva tumoral.
Con estos procedimientos normalmente se logra la curación 2.
El principal riesgo de la intervención es romper la cortical y la transformación de la lesión en fractura 2,4,7.
Se puede hacer resección quirúrgica de forma profiláctica si se encuentran en vecindad de un vaso, si impiden el movimiento articular normal, en los casos de fracturas o si existe sospecha fundada de transformación maligna2.
El pronóstico generalmente es favorable en los niños, pues cesan con el crecimiento y la madurez ósea.
Alrededor de un 1,8% de los osteocondromas recidivan debido al potencial de crecimiento.
Esta recurrencia consiste en un nuevo osteocondroma, aunque en ocasiones se trata de un condrosarcoma de bajo grado1,2.
El seguimiento de estos pacientes varía de individuo a individuo, dependiendo de la extensión de la enfermedad, el tamaño y la localización del tumor, la respuesta al tratamiento, la edad, el estado general del niño y los avances en el tratamiento.
Las terapias biológicas podrán ser posibles en el futuro1,2.
Bibliografía
1.Dickey IE. Solitary Osteochondroma. E-Medicine & Medscape [Internet] [actualizado el 15/09/2009; consultado el 14/02/2010]. Disponible en http://emedicine.medscape.com/article/1256477-overview
2.Khan AN. Osteochondroma and Osteochondromatosis. E-Medicine & Medscape [Internet] [actualizado el 19/10/2009; consultado el 14/02/2010].
Disponible en http://emedicine.medscape.com/article/392546-overview
3.Staals EL, Bacchini P, Mercuri M, Bertoni F. Dedifferentiated chondrosarcomas arising in preexisting osteochondromas. J Bone Joint Surg Am. 2007;89:987.
4.Florez B, Mönckeberg J, Castillo G, Beguiristain J. Solitary ostechondroma long-term follow-up. J. Pediatr Orthop B. 2008;17(2):91-4.
5.Mavrogenis AF, Papagelopoulos PJ, Soucacos PN. Skeletal ostechondromas revisited. Orthopedics. 2008;31(10).
6.Sepúlveda M. Tumores formadores de cartílago: clínica y tratamiento. Medwave [Internet] [actualizado en 11/2003; consultado el 14/02/ 2010]. Disponible en www.medwave.cl/cursos/Tumores/noviembre2003/3.act.
7.Murphey MD, Chol JJ, Kransdorf MJ, Flemming DJ, Gannon FH. Imaging of osteochondroma: Variants and complications with radiologic pathologic correlation.
Radiographics. 2000;20:1407-34.
8.Children´s Hospital Boston. Ostechondroma (exostosis). [Internet] [consultado el 14/02/ 2010]. Disponible en www.childrenshospital.org/az/Site1079/mainpageS1079P0.html
9.Ortega R, Fernández ME, Gómez I. Masa poplítea asociada a osteocondroma. An Pediatr (Barc). 2005;63(2):185-6.
Exostosis tibial: osteocondroma
Autores:
Parada López R a, Montano Navarro E b, Lafraya Puente AL c, Rodríguez Ortega M d
a Pediatra. CS Pinto y Getafe. Servicio Madrileño de Salud, Área 10. Madrid. España.
b Pediatra. CS Pinto y Getafe. Servicio Madrileño de Salud, Área 10. Madrid. España.
c Pediatra. CS Pinto y Getafe. Servicio Madrileño de Salud, Área 10. Madrid. España.
d Cirugía General y Aparato Digestivo. Hospital Infanta Cristina. Parla, Madrid. España.
Correspondencia: R Parada. Correo electrónico: rebecaparada@hotmail.com
Referencia para citar este artículo:
Parada López R, Montano Navarro E, Lafraya Puente AL, Rodríguez Ortega M.
Exostosis tibial: osteocondroma. Rev Pediatr Aten Primaria. 2010;12:255-261.
Resumen:
El osteocondroma o exostosis cartilaginosa es el tumor óseo más frecuente, representando el 10-15% de la totalidad. Alrededor del 3% de la población lo padece.
Es un tumor propio de individuos jóvenes con ligero predominio en varones.
Puede ser solitario o múltiple, formando parte del síndrome de exostosis múltiple hereditaria.
Suele ser un hallazgo accidental, normalmente asintomático, cuya localización más frecuente es la rodilla, aunque puede aparecer en otras localizaciones.
El diagnóstico se realiza por imagen radiográfica, que suele ser patognomónica, y el tratamiento definitivo consiste en la extirpación quirúrgica.
Introducción
Los osteocondromas son lesiones benignas formadoras de cartílago.
Suponen entre el 20-50% de los tumores benignos y entre el 10-20% de todos los tumores primarios del hueso; alrededor del 3% de la población lo padece1-3.
Son de origen cartilaginoso y se conforman tras la separación de un fragmento desde el cartílago epifisario.
Pueden ser solitarios o múltiples, y pueden aparecer espontáneamente o tras un traumatismo. Han sido descritos en prácticamente todos los huesos del esqueleto, pero tienen predilección por la metáfisis de huesos largos.
En este artículo planteamos el caso de un paciente de 13 años de edad que acude a nuestra consulta por presentar una excrecencia a nivel tibial secundaria a este tumor de origen óseo confirmado mediante radiodiagnóstico.
Caso clínico
Varón de 13 años, sin antecedentes personales de interés, que acude a la consulta del centro de salud porque desde hace unos meses ha notado una lesión en la pierna izquierda que no ocasiona ninguna sintomatología y no ha variado de tamaño.
Refiere un posible traumatismo sobre dicha zona 2 años antes tras una caída con patines.
Presenta un buen estado general y se encuentra asintomático.
Se palpa prominencia de aproximadamente 1,5-2 cm de consistencia ósea alrededor de 3-4 cm por encima del maléolo interno izquierdo.
No se objetiva inflamación local, hematoma ni dolor a la palpación.
Tampoco existen alteraciones neurovasculares distales ni impotencia funcional.
Se solicita una radiografía simple de la pierna afecta, anteroposterior y lateral, donde se visualiza una lesión de carácter óseo.
El informe radiológico da el diagnóstico:
“La radiografía anteroposterior y lateral del tobillo muestra un relieve óseo bien delimitado, a expensas de la cortical, sin aparente afectación de partes blandas, en cara medial de tercio distal de la tibia izquierda compatible con osteocondroma”.
Dado que se trata de una masa asintomática y se ha mantenido estable conservando su tamaño, se decide seguir la evolución en la consulta y ante posibles cambios realizar prueba de imagen y/o citarlo en consultas de traumatología para revisión.
Discusión
Nos encontramos ante un caso de osteocondroma, exostosis ósea recubierta de cartílago que se ubica en la superficie externa del hueso.
Es un tumor propio de individuos muy jóvenes; de hecho, el 70% de las lesiones osteocondrales se encuentra en las 2 primeras décadas de la vida2,4.
Tiene un ligero predominio en varones, con una proporción de 3/12.
Los osteocondromas se forman tras la separación de un fragmento desde el cartílago epifisario, que se hernia a través del periostio que envuelve el platillo de crecimiento.
El desarrollo posterior de este fragmento cartilaginoso y su osificación endocondral dará lugar a la exostosis recubierta de cartílago que se proyecta hacia la superficie ósea.
El cartílago extruido sigue el proceso normal de osificación, tiene un espesor menor de 1 cm y es histológicamente normal5.
Los osteocondromas pueden ser solitarios o múltiples1,4, siendo estos últimos constituyentes de la exostosis hereditaria múltiple1,3,6.
En nuestro paciente sólo se encontró una lesión y no había habido ninguna otra persona en su familia con un cuadro similar.
Algunos adolescentes refieren un antecedente de traumatismo o ejercicio vigoroso previo a la aparición del mismo9.
La exostosis puede desarrollarse en cualquier hueso del esqueleto, mostrando preferencia por las zonas metafisarias vecinas a los cartílagos más fértiles de los huesos largos: distal del fémur, proximal de la tibia, distal del radio, etc. La ubicación más frecuente es la rodilla2,4-6.
En ocasiones se desarrollan en huesos de la pelvis, escápula y costillas, tratándose de masas sésiles de pedículo corto. Es raro que afecten a los huesos cortos de manos y pies.
La lesión que comienza precozmente causa trastornos de crecimiento, con acortamiento y deformidad de la extremidad afectada.
Sin embargo, habitualmente es asintomática y se diagnostica como un hallazgo casual1, como el caso que nos ocupa.
Las deformidades esqueléticas y estéticas causadas por los osteocondromas subyacentes son la forma característica de presentación.
Igualmente, pueden producir compresión extrínseca sobre las estructuras óseas, articulares, nerviosas, musculares y ligamentosas vecinas.
También pueden producir compresión de vísceras, como ocurre en los derrames pleurales causados por los osteocondromas costales.
En las zonas de fricción ósea se suelen desarrollar bolsas sinoviales.
Las fracturas son infrecuentes y se producen principalmente en la región de la rodilla2,4,5.
Finalmente, lo más importante a tener en cuenta es que si un osteocondroma duele, además hay que descartar su posible transformación maligna (condrosarcoma)2,3,7.
El riesgo de transformación sarcomatosa en la exostosis solitaria es aproximadamente del 1%, pero en la forma múltiple hereditaria el riesgo se acerca al 10% debido a las numerosas lesiones3. Los datos que nos inducen a pensar que existe una transformación maligna son:
1) la lesión se hace sintomática de repente (duele) o empieza a crecer rápidamente;
2) la cubierta cartilaginosa es más gruesa de 1 cm en un adulto (en el niño puede ser de 2-3 cm de espesor) o con un diámetro máximo mayor de 5 cm;
3) el aumento súbito o marcado en la captación en la gammagrafía ósea en un adulto (en la madurez esquelética lo normal es que esté latente); y
4) la confirmación por la TC (tomografía computadorizada) o RM (resonancia magnética) de una masa de tejidos blandos o desplazamiento de un paquete neurovascular mayor4.
Normalmente los grandes osteocondromas suelen producir desplazamiento de los vasos vecinos. Las complicaciones vasculares de los osteocondromas incluyen: pseudoaneurismas, oclusiones arteriales y venosas y fístulas arteriovenosas.
El 90% de las complicaciones vasculares afectan directamente a las arterias, siendo los pseudoaneurismas la patología más frecuente (63,9%)3-5.
En cuanto al diagnóstico, la prueba de imagen más usada es la radiografía simple.
La apariencia radiográfica de una exostosis es la de una lesión aplanada, sésil o pediculada3-4 (imagen radiológica patognomónica).
Actualmente, la ecografía es el método más accesible en nuestro medio.
Es útil en la valoración del casquete cartilaginoso y además tiene la ventaja de que en los casos de existencia de pseudoaneurisma, muestra con facilidad la presencia de una masa compleja con flujo en su interior y dependiente de una gran arteria.
Otros métodos de imagen son:
la TC (define estructuras óseas anómalas y su relación con complicaciones vasculares mediante la inyección de contraste endovenoso y es de gran utilidad en el estudio del osteocondroma que se origina en hombro, pelvis, columna vertebral y escápula) y la RM, prueba de elección ante la sospecha de malignización. Permite detectar la continuidad entre el hueso medular y cortical del tumor y el hueso afecto. Además el casquete cartilaginoso es hiperintenso en secuencia T21-2.
Cuando en la consulta de Atención Primaria nos encontramos un caso como éste, debemos llevar a cabo un diagnóstico diferencial, aunque su presentación clínica no suele presentar problemas para el diagnóstico. Debemos diferenciarlo principalmente de la exostosis cartilaginosa múltiple y el osteosarcoma parostal.
La exostosis cartilaginosa múltiple es una enfermedad congénita caracterizada por tumores óseos que se distribuyen por casi todo el esqueleto, descrita ya en 1891 por Bessel-Hagen.
Tiene una herencia autosómica dominante.
Se han identificado tres loci en relación con la enfermedad:
EXT1, en el cromosoma 8q23-q24;
EXT2 en el 11p11-p12 y
EXT3 en el brazo corto del cromosoma 19.
Alrededor de 2/3 de los pacientes tienen antecedentes familiares de la enfermedad.
Esta forma múltiple tiene una prevalencia estimada entre el 1/1.000 y el 1/50.000, dependiendo de la zona geográfica que se estudie7.
Se debe buscar esta entidad en pacientes con estatura corta, achatamiento de radio y deformidad angular de los miembros inferiores, signos que no están presentes en nuestro paciente.
El osteosarcoma parostal puede presentarse como una exostosis sintomática que aumenta de tamaño en los adultos.
Está compuesto de tejidos fibro-óseos densos que no están presentes en el osteocondroma.
Con respecto al tratamiento del osteocondroma, existen dos opciones;
a.- por un lado el conservador, en aquellos casos que resultan asintomáticos y se mantienen estables y,
b.-por otro, el quirúrgico, indicado en las formas sintomáticas de la infancia y el crecimiento continuado del osteocondroma después de la madurez del esqueleto (sospecha de malignización)4.
El tratamiento definitivo consiste en una resección simple o ampliada.
Si es pediculado se hace la resección simple hasta la base.
Cuando es sésil se hace una resección ampliada sacando una zona de cortical y completando con curetaje 6, extirpando siempre la exostosis, su cobertura cartilaginosa y el periostio, ya que los restos de éste y del pericondrio pueden dar lugar a una recidiva tumoral.
Con estos procedimientos normalmente se logra la curación 2.
El principal riesgo de la intervención es romper la cortical y la transformación de la lesión en fractura 2,4,7.
Se puede hacer resección quirúrgica de forma profiláctica si se encuentran en vecindad de un vaso, si impiden el movimiento articular normal, en los casos de fracturas o si existe sospecha fundada de transformación maligna2.
El pronóstico generalmente es favorable en los niños, pues cesan con el crecimiento y la madurez ósea.
Alrededor de un 1,8% de los osteocondromas recidivan debido al potencial de crecimiento.
Esta recurrencia consiste en un nuevo osteocondroma, aunque en ocasiones se trata de un condrosarcoma de bajo grado1,2.
El seguimiento de estos pacientes varía de individuo a individuo, dependiendo de la extensión de la enfermedad, el tamaño y la localización del tumor, la respuesta al tratamiento, la edad, el estado general del niño y los avances en el tratamiento.
Las terapias biológicas podrán ser posibles en el futuro1,2.
Bibliografía
1.Dickey IE. Solitary Osteochondroma. E-Medicine & Medscape [Internet] [actualizado el 15/09/2009; consultado el 14/02/2010]. Disponible en http://emedicine.medscape.com/article/1256477-overview
2.Khan AN. Osteochondroma and Osteochondromatosis. E-Medicine & Medscape [Internet] [actualizado el 19/10/2009; consultado el 14/02/2010].
Disponible en http://emedicine.medscape.com/article/392546-overview
3.Staals EL, Bacchini P, Mercuri M, Bertoni F. Dedifferentiated chondrosarcomas arising in preexisting osteochondromas. J Bone Joint Surg Am. 2007;89:987.
4.Florez B, Mönckeberg J, Castillo G, Beguiristain J. Solitary ostechondroma long-term follow-up. J. Pediatr Orthop B. 2008;17(2):91-4.
5.Mavrogenis AF, Papagelopoulos PJ, Soucacos PN. Skeletal ostechondromas revisited. Orthopedics. 2008;31(10).
6.Sepúlveda M. Tumores formadores de cartílago: clínica y tratamiento. Medwave [Internet] [actualizado en 11/2003; consultado el 14/02/ 2010]. Disponible en www.medwave.cl/cursos/Tumores/noviembre2003/3.act.
7.Murphey MD, Chol JJ, Kransdorf MJ, Flemming DJ, Gannon FH. Imaging of osteochondroma: Variants and complications with radiologic pathologic correlation.
Radiographics. 2000;20:1407-34.
8.Children´s Hospital Boston. Ostechondroma (exostosis). [Internet] [consultado el 14/02/ 2010]. Disponible en www.childrenshospital.org/az/Site1079/mainpageS1079P0.html
9.Ortega R, Fernández ME, Gómez I. Masa poplítea asociada a osteocondroma. An Pediatr (Barc). 2005;63(2):185-6.
martes, 17 de agosto de 2010
¿Que son los Osteocondromas en el hombro?
http://www.thehouseofblogs.com/articulo/revistas_gratuitas__dolor_de_hombro_en_paciente-121720.html
Revistas gratuitas : Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico
Fuente: La Medicina Natural y Remedios Naturales.16 del 8 de 2010
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico
http://www.portalesmedicos.com/publicaciones/articles/2089/1/Dolor-de-hombro-en-paciente-con-exostosis-multiple-A-proposito-de-un-caso-clinico.html
Dolor de hombro en paciente con exóstosis múltiple. A propósito de un caso clínico
Mesa Rivero M. E. (MIR),
Rodríguez de la Cueva J. M. (FEA),
González Herranz J. (FEA),
Angulo Gutiérrez J. (FEA).
Servicio de Cirugía Ortopédica y Traumatología. Hospital Universitario de Valme. Sevilla (España).
Dolor de hombro en paciente con exóstosis múltiple. A propósito de un caso.
1. Resumen:
Objetivo: La exóstosis hereditaria múltiple es una enfermedad autosómica dominante caracterizada por la aparición de osteocondromas, de predominio metafisario de los huesos largos (1) y que presentan un recubrimiento cartilaginoso benigno.
Los osteocondromas crecen y se desarrollan en la primera década de la vida. Suelen ser asintomáticos, aunque pueden ocasionar dolor local y compresión de estructuras vasculonerviosas adyacentes o degeneración maligna hacia condrosarcoma, en un 0.5-5% de los casos (2).
Material y método: Presentamos el caso de un paciente de 12 años de edad, afectado de exóstosis múltiple, que acude a nuestras consultas con dolor en hombro izquierdo.
Se realizó tratamiento mediante exéresis de un pedículo tumoral, desapareciendo la clínica y evolucionando favorablemente.
Conclusión: En el estudio anatomopatológico se diagnóstico de osteocondromas múltiples. Consideramos la importancia del caso debido al potencial de malignización de estas lesiones así como la necesidad de seguimiento genésico de los pacientes.
2. Abstract:
Hereditary multiple exostoses is an autosomal dominant inheritance pattern, characteriszed by benign cartilage-capped bone tumours (osteochondromas) that grow outward from the metaphyses of the long bones.
Osteochondromas develop and increase in size in the first decade of life.
The majority are asymptomatic, they may even cause pain and impingement on adjacents nerves and vessels or malignant transformation of osteochondroma towards chondrosarcoma (0.5-5%).
We present a case of a 12 year-old patient with multiple exostoses and left shoulder pain.
We resected a humeral tumour.
The symptoms disappeared, with a good clinic evolution.
Anatomicopathologists carried out a diagnosis of multiple osteochondromas.
We consider the relevance of this case because of the malignant degeneration.
Patients should be well instructed and followed-up for detection of malignancy.
Key words: osteochondromas, multiple exostoses, chondrosarcomas.
3. Introducción:
La exóstosis hereditaria múltiple constituye una entidad caracterizada por el desarrollo de 20 ó más tumores de localización metafisaria y que, típicamente, presentan un recubrimiento cartilaginoso (1).
Su prevalencia es del 1 entre 50.000 habitantes, siendo más frecuente en los varones (2).
En su origen está implicada la mutación de los genes EXT1 Y EXT2, presentando un patrón de herencia autosómica dominante (3).
Los osteocondromas crecen y se desarrollan en la primera década de la vida, con un número de localizaciones variables entre los individuos de una misma familia.
Se localizan principalmente en la metáfisis de los huesos largos, sobre todo próximos a la rodilla. La mayoría son asintomáticos aunque, en una menor proporción pueden producir dolor, compresión de estructuras adyacentes, deformidad, problemas funcionales y degeneración maligna hacia condrosarcoma, justificando dicha sintomatología la exéresis tumoral (2).
El diagnóstico se realiza mediante la clínica del paciente, apoyado por la pruebas de imagen (4) y, a ser posible, complementándose con la historia familiar y el estudio genético de la misma.
El tratamiento quirúrgico mediante la exéresis tumoral está indicado cuando aparecen complicaciones derivadas por un excesivo crecimiento ó por la malignización.
Los osteocondromas son lesiones benignas, dependiendo así el pronóstico de la degeneración maligna hacia condrosarcoma (5).
El patrón de transmisión es autosómico dominante, con una penetrancia del 100%; así, en caso de aislarse el gen afectado, existe la posibilidad de realizar el diagnóstico prenatal (2).
4. Material y método:
Paciente varón de 12 años de edad, que acude al Servicio de Urgencias de Traumatología al presentar dolor en hombro izquierdo de inicio súbito y sin traumatismo previo.
No existen antecedentes personales de interés.
Como antecedentes familiares refiere que 2 hermanos de su madre han sido diagnosticados de exostosis múltiple, sin datos de degeneración maligna en ninguno de ellos.
A la exploración presenta una talla baja para su edad, con extremidades relativamente cortas con respecto al tronco, así como prominencias óseas a nivel de las rodillas, muñecas y hombros.
Consideramos como significativo una limitación de la prono-supinación en ambos extremidades superiores. A la palpación refiere dolor en la zona deltoidea de miembro superior izquierdo y limitación de las rotaciones en ambas articulaciones coxo-femorales.
Realizamos un mapa óseo, hallándose lesiones óseas de predominio metafisario en las tibias, fémures y húmeros, compatibles con el diagnostico de osteocondromatosis.
Destaca la presencia de una fractura del pedículo de un osteocondroma en la metáfisis del húmero izquierdo, así como de un ensanchamiento de la metáfisis del húmero derecho debido a la presencia de varios osteocondromas.
Ampliamos el estudio realizando una TAC de la extremidad proximal de ambos húmeros, donde lo más destacado es la fractura del pedículo óseo antes mencionado en el húmero izquierdo, y la presencia de múltiples osteocondromas en la región proximal de húmero y escápula derecha, llamando especialmente la atención la presencia de un osteocondroma con mayor densidad ósea, en la cara interna de la escápula, que separa a esta de la pared torácica.
En la RMN no se observan, signos inflamatorios, formación de alguna colección, o otras alteraciones en las partes blandas que nos hagan sospechar malignización del proceso.
Realizamos la exéresis transpedicular de 2 osteocondromas en escápula derecha; así como la extirpación, por vía deltopectoral, de 3 tumores en el extremo proximal del húmero izquierdo.
El material obtenido es remitido como material de biopsia para estudio anatomo-patológico.
5. Resultado:
El resultado del estudio anatomopatológico, reveló la presencia de lesiones de tipo osteocartilaginoso, con centro compuesto por material osteoide y recubrimiento superficial de cartílago. Siendo diagnosticado de osteocondromas.
El paciente fue dado de alta con tratamiento analgésicos habitual y, realizando controles periódicos para el seguimiento de su proceso en régimen ambulatorio y evitar una demora en el diagnostico de una posible degeneración maligna.
6. Discusión:
La exóstosis múltiple es una enfermedad que presenta típicamente tumores (osteocondromas) de predominio metafisario y recubiertos por cartílago, apareciendo sobre todo en los huesos largos (1).
La prevalencia es variable, siendo de 1 cada 100.000 individuos en Europa y de 1/50.000 en el estado de Washington (1).
Son más frecuentes en el varón, existiendo una relación de 1.5: 1, entre hombres y mujeres.
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico .2
Los osteocondromas solitarios son 6 veces más frecuentes que la exóstosis múltiple.
El 62% de los pacientes afectados, presentan historia familiar positiva para la enfermedad (3).
En nuestro caso, el paciente presenta un antecedente familiar, 2 tíos varones por vía materna, existiendo la sospecha de afectación familiar en 3 generaciones anteriores.
La historia familiar sugiere un patrón de herencia autosómico dominante, con una penetrancia de casi el 100%.
Parece que en el desarrollo de la enfermedad se ha implicado la mutación del gen EXT, que actúa como un gen supresor tumoral.
Dicho gen actúa como productor de glicosiltranferasas, relacionadas con la síntesis de un recubrimiento celular de heparan sulfato, afectando así la difusión del morfogen Hedgehog (5).
Se ha identificado una mutación cromosómica en tres localizaciones distintas:
EXT 1 (cromosoma 8, locus 8q224.1);
EXT 2 (cromosoma 11, en la región pericéntrica);
EXT 3 (cromosoma 19, locus 19 p),
siendo la mutación del gen EXT 1 la más frecuente cuando existe historia familiar de la enfermedad (1).
El proceso de malignización está representado por una inestabilidad cromosómica, previamente demostrada por un alto porcentaje de pérdida de heterocigosidad, y que implica a varios loci en un amplio rango del DNA.
En el caso presentado no hemos realizado estudio genético.
La proporción de individuos que presentan sintomatología clínica incrementa con la edad, siendo bajo al nacer (5%), aumentado con edad (a los 12 años el 96% ha presentado alguna clínica).
En la edad puberal y, coincidiendo con el cierre del cartílago de crecimiento, es cuando los osteocondromas dejan de crecer (1).
Los síntomas principales de la exóstosis múltiple son:
a.- el dolor,
b.- la limitación funcional de alguna articulación,
c.- es frecuente el acortamiento del cúbito que ocasiona una incurvación del radio (39-60% de los casos),
d.- así como la dismetría de las extremidades (10-50%), y
e.- las deformidades angulares en varo ó valgo de la rodilla (8-33%),
f.- como de los tobillos (2-54%).
g.- Con frecuencia se asocia talla baja ¿rizomélica? (37-44%).
En menor proporción podemos encontrarnos con otras manifestaciones osteoarticulares como bursitis, compresión de estructuras vasculonerviosas, fracturas del pedículo, y dolores difusos generalizados. (3)
La complicación más importante es la degeneración maligna en condrosarcoma, que se presenta entre un 0.5-5% de los casos, y que suele manifestarse como dolor y aumento de la masa tumoral; así como engrosamiento de la capa cartilaginosa que recubre al tumor en más de 1 cm..
La degeneración hacia otras lesiones malignas como fibrosarcoma, fribrohistiocitoma maligno y osteosarcoma, se presenta con menor frecuencia (5).
Los osteocondromas son raros en el esqueleto axial, habiéndose descrito algunos casos de compresión medular a nivel de C1, C2 y C5.
Si sospechamos que un paciente padece de exóstosis múltiple, debemos realizar:
1.- detallado estudio de la historia familiar,
2.- un estudio radiográfico e histológico tumoral y,
3.- cuando sea posible, un estudio genético.
El estudio radiográfico se inicia mediante un mapa óseo, siendo necesario una TAC para poder valorar con exactitud las lesiones y esclarecer el origen de dichas lesiones (2).
Con el TAC podemos realizar un estudio adecuado de la anatomía ósea regional y de las fracturas de las lesiones exofíticas (5).
La RMN con contraste nos permite identificar tanto la morfología del cartílago que recubre al tumor, como de las partes blandas peritumorales, que nos podrá indicar una posible degeneración maligna (5).
En nuestro caso realizamos un mapa óseo al paciente, así como una TAC de la extremidad superior de ambos húmeros debido a los hallazgos previos radiográficos, con la intención de identificar la extensión y crecimiento de los osteocondromas de estas regiones.
Las imágenes de la RNM nos ayudo a descartar la malignización de las lesiones más bizarras.
Debemos realizar un diagnóstico diferencial con:
a.- la Displasia Hemimélica Epifisaria (enfermedad de Trévor) y
b.- la metacondromatosis, enfermedades que no guardan relación con una mutación del gen EXT;
c.- así como diferenciarlo de la enfermedad de Mafucci y Ollier, cuyo patrón de presentación es distinto (3).
El manejo de la exóstosis múltiple, así como su tratamiento, es muy variado.
Cuando dichas lesiones causan dolor, compresión de estructuras adyacentes, deformidad estética ó limitación funcional, está indicado el tratamiento quirúrgico, realizando la exéresis de los osteocondromas que causan estas complicaciones (3).
El tratamiento de la enfermedad del antebrazo es controvertido.
Akita et al. en una serie de 23 pacientes, concluyeron que la osteotomía y el alargamiento de los huesos del antebrazo no era muy beneficiosa.
Así, el procedimiento más adecuado, sería la exéresis del tumor, mejorando la movilidad cuando la lesión afecta al extremo distal del cúbito (3).
Hay que advertir al paciente y sus familiares que debe ser revisado de forma periódica y que en el caso que surja algún cambio en la evolución que solicite una consulta médica de forma inmediata.
Tras la pubertad, los osteocondromas no deben de aumentar de tamaño, así una vez alcanzada la madurez esquelética, se recomienda realizar controles anuales, aunque aún no se ha demostrado la eficacia de dicho seguimiento (3).
En el caso de sufrir la malignización de un osteocondroma, está indicada la resección en bloque, incluyendo la pseudocápsula tumoral y márgenes libres de tumor, ya que así podríamos lograr un buen resultado local y a largo plazo.
A nuestro paciente se le realizó:
1.- exéresis transpedicular del tumor de escápula derecha; y
2.- la exéresis del tumor sésil localizado en metáfisis de húmero izquierdo, cuya rotura era la responsable del dolor que presentaba en hombro izquierdo.
Los osteocondromas son tumores benignos y no afectan la esperanza de vida (3).
El riesgo de malignización hacia osteosarcoma es aproximadamente del 1% y, en caso de producirse, el pronóstico dependerá del grado histológico, siendo:
a.- la supervivencia a los 10 años del 83% para el grado I y
b.- del 29% para el grado III (3).
7. Bibliografía:
1. Gupta PP, Agarwal D. A middle-aged manwith persisting chest opacity and permanent bony swelling. CMAJ 2006 Nov 15 ; 31 (24): E920-4
2. Manish Chadha, MS; Arun Pal Singh, MS. Secondary chondrosarcoma of the cuboid bone in a patient with multiple exostosis. Can J Surg, 2008.Vol. 51 (1).
3. Judith VMG Bovée. Multiple osteochondromas. Orphanet Journal of Rare Diseases 2008, 3:3
4. Carolyn M. Sofka, MD& Gregory R. Saboeiro, MD& Robert Schneider, MD. Multiple Hereditary Exostoses. HSSJ (2005) 1:49–51.
5. J V M G Bovée, R J B Sakkers, M J A Geirnaerdt, A H M Taminiau, P C W Hogendoorn. Intermediate grade osteosarcoma and chondrosarcoma
6. Arising in an osteochondroma. A case report of a patient with hereditary multiple exostoses.J Clin Pathol 2002;55:226–229
Fuente: Portalesmadicos.com
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico
Blogalaxia Tags: medicina medicina alternativa tratamientos naturales remedios caseros remedios para
Webs Recomendadas: remedios y tratamientos naturalesmedicina natural y tratamientostratamientos naturalesTratamientos Medicinales Gratis
Revistas gratuitas : Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico
Fuente: La Medicina Natural y Remedios Naturales.16 del 8 de 2010
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico
http://www.portalesmedicos.com/publicaciones/articles/2089/1/Dolor-de-hombro-en-paciente-con-exostosis-multiple-A-proposito-de-un-caso-clinico.html
Dolor de hombro en paciente con exóstosis múltiple. A propósito de un caso clínico
Mesa Rivero M. E. (MIR),
Rodríguez de la Cueva J. M. (FEA),
González Herranz J. (FEA),
Angulo Gutiérrez J. (FEA).
Servicio de Cirugía Ortopédica y Traumatología. Hospital Universitario de Valme. Sevilla (España).
Dolor de hombro en paciente con exóstosis múltiple. A propósito de un caso.
1. Resumen:
Objetivo: La exóstosis hereditaria múltiple es una enfermedad autosómica dominante caracterizada por la aparición de osteocondromas, de predominio metafisario de los huesos largos (1) y que presentan un recubrimiento cartilaginoso benigno.
Los osteocondromas crecen y se desarrollan en la primera década de la vida. Suelen ser asintomáticos, aunque pueden ocasionar dolor local y compresión de estructuras vasculonerviosas adyacentes o degeneración maligna hacia condrosarcoma, en un 0.5-5% de los casos (2).
Material y método: Presentamos el caso de un paciente de 12 años de edad, afectado de exóstosis múltiple, que acude a nuestras consultas con dolor en hombro izquierdo.
Se realizó tratamiento mediante exéresis de un pedículo tumoral, desapareciendo la clínica y evolucionando favorablemente.
Conclusión: En el estudio anatomopatológico se diagnóstico de osteocondromas múltiples. Consideramos la importancia del caso debido al potencial de malignización de estas lesiones así como la necesidad de seguimiento genésico de los pacientes.
2. Abstract:
Hereditary multiple exostoses is an autosomal dominant inheritance pattern, characteriszed by benign cartilage-capped bone tumours (osteochondromas) that grow outward from the metaphyses of the long bones.
Osteochondromas develop and increase in size in the first decade of life.
The majority are asymptomatic, they may even cause pain and impingement on adjacents nerves and vessels or malignant transformation of osteochondroma towards chondrosarcoma (0.5-5%).
We present a case of a 12 year-old patient with multiple exostoses and left shoulder pain.
We resected a humeral tumour.
The symptoms disappeared, with a good clinic evolution.
Anatomicopathologists carried out a diagnosis of multiple osteochondromas.
We consider the relevance of this case because of the malignant degeneration.
Patients should be well instructed and followed-up for detection of malignancy.
Key words: osteochondromas, multiple exostoses, chondrosarcomas.
3. Introducción:
La exóstosis hereditaria múltiple constituye una entidad caracterizada por el desarrollo de 20 ó más tumores de localización metafisaria y que, típicamente, presentan un recubrimiento cartilaginoso (1).
Su prevalencia es del 1 entre 50.000 habitantes, siendo más frecuente en los varones (2).
En su origen está implicada la mutación de los genes EXT1 Y EXT2, presentando un patrón de herencia autosómica dominante (3).
Los osteocondromas crecen y se desarrollan en la primera década de la vida, con un número de localizaciones variables entre los individuos de una misma familia.
Se localizan principalmente en la metáfisis de los huesos largos, sobre todo próximos a la rodilla. La mayoría son asintomáticos aunque, en una menor proporción pueden producir dolor, compresión de estructuras adyacentes, deformidad, problemas funcionales y degeneración maligna hacia condrosarcoma, justificando dicha sintomatología la exéresis tumoral (2).
El diagnóstico se realiza mediante la clínica del paciente, apoyado por la pruebas de imagen (4) y, a ser posible, complementándose con la historia familiar y el estudio genético de la misma.
El tratamiento quirúrgico mediante la exéresis tumoral está indicado cuando aparecen complicaciones derivadas por un excesivo crecimiento ó por la malignización.
Los osteocondromas son lesiones benignas, dependiendo así el pronóstico de la degeneración maligna hacia condrosarcoma (5).
El patrón de transmisión es autosómico dominante, con una penetrancia del 100%; así, en caso de aislarse el gen afectado, existe la posibilidad de realizar el diagnóstico prenatal (2).
4. Material y método:
Paciente varón de 12 años de edad, que acude al Servicio de Urgencias de Traumatología al presentar dolor en hombro izquierdo de inicio súbito y sin traumatismo previo.
No existen antecedentes personales de interés.
Como antecedentes familiares refiere que 2 hermanos de su madre han sido diagnosticados de exostosis múltiple, sin datos de degeneración maligna en ninguno de ellos.
A la exploración presenta una talla baja para su edad, con extremidades relativamente cortas con respecto al tronco, así como prominencias óseas a nivel de las rodillas, muñecas y hombros.
Consideramos como significativo una limitación de la prono-supinación en ambos extremidades superiores. A la palpación refiere dolor en la zona deltoidea de miembro superior izquierdo y limitación de las rotaciones en ambas articulaciones coxo-femorales.
Realizamos un mapa óseo, hallándose lesiones óseas de predominio metafisario en las tibias, fémures y húmeros, compatibles con el diagnostico de osteocondromatosis.
Destaca la presencia de una fractura del pedículo de un osteocondroma en la metáfisis del húmero izquierdo, así como de un ensanchamiento de la metáfisis del húmero derecho debido a la presencia de varios osteocondromas.
Ampliamos el estudio realizando una TAC de la extremidad proximal de ambos húmeros, donde lo más destacado es la fractura del pedículo óseo antes mencionado en el húmero izquierdo, y la presencia de múltiples osteocondromas en la región proximal de húmero y escápula derecha, llamando especialmente la atención la presencia de un osteocondroma con mayor densidad ósea, en la cara interna de la escápula, que separa a esta de la pared torácica.
En la RMN no se observan, signos inflamatorios, formación de alguna colección, o otras alteraciones en las partes blandas que nos hagan sospechar malignización del proceso.
Realizamos la exéresis transpedicular de 2 osteocondromas en escápula derecha; así como la extirpación, por vía deltopectoral, de 3 tumores en el extremo proximal del húmero izquierdo.
El material obtenido es remitido como material de biopsia para estudio anatomo-patológico.
5. Resultado:
El resultado del estudio anatomopatológico, reveló la presencia de lesiones de tipo osteocartilaginoso, con centro compuesto por material osteoide y recubrimiento superficial de cartílago. Siendo diagnosticado de osteocondromas.
El paciente fue dado de alta con tratamiento analgésicos habitual y, realizando controles periódicos para el seguimiento de su proceso en régimen ambulatorio y evitar una demora en el diagnostico de una posible degeneración maligna.
6. Discusión:
La exóstosis múltiple es una enfermedad que presenta típicamente tumores (osteocondromas) de predominio metafisario y recubiertos por cartílago, apareciendo sobre todo en los huesos largos (1).
La prevalencia es variable, siendo de 1 cada 100.000 individuos en Europa y de 1/50.000 en el estado de Washington (1).
Son más frecuentes en el varón, existiendo una relación de 1.5: 1, entre hombres y mujeres.
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico .2
Los osteocondromas solitarios son 6 veces más frecuentes que la exóstosis múltiple.
El 62% de los pacientes afectados, presentan historia familiar positiva para la enfermedad (3).
En nuestro caso, el paciente presenta un antecedente familiar, 2 tíos varones por vía materna, existiendo la sospecha de afectación familiar en 3 generaciones anteriores.
La historia familiar sugiere un patrón de herencia autosómico dominante, con una penetrancia de casi el 100%.
Parece que en el desarrollo de la enfermedad se ha implicado la mutación del gen EXT, que actúa como un gen supresor tumoral.
Dicho gen actúa como productor de glicosiltranferasas, relacionadas con la síntesis de un recubrimiento celular de heparan sulfato, afectando así la difusión del morfogen Hedgehog (5).
Se ha identificado una mutación cromosómica en tres localizaciones distintas:
EXT 1 (cromosoma 8, locus 8q224.1);
EXT 2 (cromosoma 11, en la región pericéntrica);
EXT 3 (cromosoma 19, locus 19 p),
siendo la mutación del gen EXT 1 la más frecuente cuando existe historia familiar de la enfermedad (1).
El proceso de malignización está representado por una inestabilidad cromosómica, previamente demostrada por un alto porcentaje de pérdida de heterocigosidad, y que implica a varios loci en un amplio rango del DNA.
En el caso presentado no hemos realizado estudio genético.
La proporción de individuos que presentan sintomatología clínica incrementa con la edad, siendo bajo al nacer (5%), aumentado con edad (a los 12 años el 96% ha presentado alguna clínica).
En la edad puberal y, coincidiendo con el cierre del cartílago de crecimiento, es cuando los osteocondromas dejan de crecer (1).
Los síntomas principales de la exóstosis múltiple son:
a.- el dolor,
b.- la limitación funcional de alguna articulación,
c.- es frecuente el acortamiento del cúbito que ocasiona una incurvación del radio (39-60% de los casos),
d.- así como la dismetría de las extremidades (10-50%), y
e.- las deformidades angulares en varo ó valgo de la rodilla (8-33%),
f.- como de los tobillos (2-54%).
g.- Con frecuencia se asocia talla baja ¿rizomélica? (37-44%).
En menor proporción podemos encontrarnos con otras manifestaciones osteoarticulares como bursitis, compresión de estructuras vasculonerviosas, fracturas del pedículo, y dolores difusos generalizados. (3)
La complicación más importante es la degeneración maligna en condrosarcoma, que se presenta entre un 0.5-5% de los casos, y que suele manifestarse como dolor y aumento de la masa tumoral; así como engrosamiento de la capa cartilaginosa que recubre al tumor en más de 1 cm..
La degeneración hacia otras lesiones malignas como fibrosarcoma, fribrohistiocitoma maligno y osteosarcoma, se presenta con menor frecuencia (5).
Los osteocondromas son raros en el esqueleto axial, habiéndose descrito algunos casos de compresión medular a nivel de C1, C2 y C5.
Si sospechamos que un paciente padece de exóstosis múltiple, debemos realizar:
1.- detallado estudio de la historia familiar,
2.- un estudio radiográfico e histológico tumoral y,
3.- cuando sea posible, un estudio genético.
El estudio radiográfico se inicia mediante un mapa óseo, siendo necesario una TAC para poder valorar con exactitud las lesiones y esclarecer el origen de dichas lesiones (2).
Con el TAC podemos realizar un estudio adecuado de la anatomía ósea regional y de las fracturas de las lesiones exofíticas (5).
La RMN con contraste nos permite identificar tanto la morfología del cartílago que recubre al tumor, como de las partes blandas peritumorales, que nos podrá indicar una posible degeneración maligna (5).
En nuestro caso realizamos un mapa óseo al paciente, así como una TAC de la extremidad superior de ambos húmeros debido a los hallazgos previos radiográficos, con la intención de identificar la extensión y crecimiento de los osteocondromas de estas regiones.
Las imágenes de la RNM nos ayudo a descartar la malignización de las lesiones más bizarras.
Debemos realizar un diagnóstico diferencial con:
a.- la Displasia Hemimélica Epifisaria (enfermedad de Trévor) y
b.- la metacondromatosis, enfermedades que no guardan relación con una mutación del gen EXT;
c.- así como diferenciarlo de la enfermedad de Mafucci y Ollier, cuyo patrón de presentación es distinto (3).
El manejo de la exóstosis múltiple, así como su tratamiento, es muy variado.
Cuando dichas lesiones causan dolor, compresión de estructuras adyacentes, deformidad estética ó limitación funcional, está indicado el tratamiento quirúrgico, realizando la exéresis de los osteocondromas que causan estas complicaciones (3).
El tratamiento de la enfermedad del antebrazo es controvertido.
Akita et al. en una serie de 23 pacientes, concluyeron que la osteotomía y el alargamiento de los huesos del antebrazo no era muy beneficiosa.
Así, el procedimiento más adecuado, sería la exéresis del tumor, mejorando la movilidad cuando la lesión afecta al extremo distal del cúbito (3).
Hay que advertir al paciente y sus familiares que debe ser revisado de forma periódica y que en el caso que surja algún cambio en la evolución que solicite una consulta médica de forma inmediata.
Tras la pubertad, los osteocondromas no deben de aumentar de tamaño, así una vez alcanzada la madurez esquelética, se recomienda realizar controles anuales, aunque aún no se ha demostrado la eficacia de dicho seguimiento (3).
En el caso de sufrir la malignización de un osteocondroma, está indicada la resección en bloque, incluyendo la pseudocápsula tumoral y márgenes libres de tumor, ya que así podríamos lograr un buen resultado local y a largo plazo.
A nuestro paciente se le realizó:
1.- exéresis transpedicular del tumor de escápula derecha; y
2.- la exéresis del tumor sésil localizado en metáfisis de húmero izquierdo, cuya rotura era la responsable del dolor que presentaba en hombro izquierdo.
Los osteocondromas son tumores benignos y no afectan la esperanza de vida (3).
El riesgo de malignización hacia osteosarcoma es aproximadamente del 1% y, en caso de producirse, el pronóstico dependerá del grado histológico, siendo:
a.- la supervivencia a los 10 años del 83% para el grado I y
b.- del 29% para el grado III (3).
7. Bibliografía:
1. Gupta PP, Agarwal D. A middle-aged manwith persisting chest opacity and permanent bony swelling. CMAJ 2006 Nov 15 ; 31 (24): E920-4
2. Manish Chadha, MS; Arun Pal Singh, MS. Secondary chondrosarcoma of the cuboid bone in a patient with multiple exostosis. Can J Surg, 2008.Vol. 51 (1).
3. Judith VMG Bovée. Multiple osteochondromas. Orphanet Journal of Rare Diseases 2008, 3:3
4. Carolyn M. Sofka, MD& Gregory R. Saboeiro, MD& Robert Schneider, MD. Multiple Hereditary Exostoses. HSSJ (2005) 1:49–51.
5. J V M G Bovée, R J B Sakkers, M J A Geirnaerdt, A H M Taminiau, P C W Hogendoorn. Intermediate grade osteosarcoma and chondrosarcoma
6. Arising in an osteochondroma. A case report of a patient with hereditary multiple exostoses.J Clin Pathol 2002;55:226–229
Fuente: Portalesmadicos.com
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico
Blogalaxia Tags: medicina medicina alternativa tratamientos naturales remedios caseros remedios para
Webs Recomendadas: remedios y tratamientos naturalesmedicina natural y tratamientostratamientos naturalesTratamientos Medicinales Gratis
Suscribirse a:
Comentarios (Atom)