http://www.europapress.es/andalucia/sevilla-00357/noticia-hospital-jaen-suma-servicios-analisis-genetico-mutaciones-causan-fribrosis-quistica-20100616184657.html
El Hospital de Jaén suma a sus servicios el análisis genético de las mutaciones que causan la fribrosis quística
JAÉN, 16 Jun. (EUROPA PRESS) -
El Complejo Hospitalario de Jaén ha incorporado en su cartera de servicios el análisis genético de las mutaciones que causan fibrosis quística en pacientes con el objetivo de promover la deteccion precoz y la instauración de un tratamiento temprano integral, que pueda mejorar tanto la supervivencia como la calidad de vida.
El test, según informó hoy el Complejo en un comunicado, se realiza en una muestra de sangre periférica en niños y bebes con problemas respiratorios asociados con manifestaciones digestivas o en aquellas personas con antecedentes familiares de esta enfermedad.
En la actualidad, y desde finales del año 2009, la Unidad de Gestión Clínica de Laboratorio del hospital jiennense está analizando las 36 mutaciones de fibrosis quística más frecuentes descrita en nuestra población.
La detección precoz de esta enfermedad mediante el estudio genético es importante, ya que permite aplicar medidas que contribuyen a reducir sus consecuencias.
Además, el estudio genético de las mutaciones causantes de fibrosis quística hace posible la realización de consejo genético.
La Fibrosis Quística es una enfermedad genética que afecta, en especial medida, al aparato respiratorio y digestivo, causando discapacidad progresiva y muerte prematura, por la acumulación de un moco espeso y pegajoso en los pulmones y tubo digestivo.
Es uno de los tipos de enfermedad pulmonar crónica más común en niños y adultos jóvenes.
Se manifiesta en edades muy tempranas y se considera una de las enfermedades hereditarias más comunes.
1 de cada 25 personas es portadora del gen defectuoso de la fibrosis quística, pudiéndola trasmitir a sus hijos si coincide con otro progenitor también portador de la misma patología.
En España afecta a 3.000 personas y a cerca de medio millar en Andalucía.
La esperanza de vida media de los pacientes de esta enfermedad es de aproximadamente 35 a 40 años.
Un diagnóstico precoz y un tratamiento adecuado aminoran los efectos de la patología y mejoran la calidad de vida.
Los afectados pueden ser diagnosticados mediante pruebas genéticas prenatales, también por 'screening' neonatal o durante la infancia temprana.
La Consejería de Salud cuenta entre las medidas sobre este tipo de dolencias realizar estudios genéticos a todos los recién nacidos como prueba de screening, tal y como se contempla en el Plan de Atención a Personas Afectadas por enfermedades raras (2008-2012).
BIOLOGÍA MOLECULAR
El Complejo Hospitalario de Jaén cuenta con una nueva Unidad de Biología Molecular que ha realizado en torno a las 5.500 determinaciones durante su 1º año de funcionamiento, una actividad que ha permitido la realización de diferentes técnicas que ayudan al diagnóstico, la prevención, elección de tratamiento y la evaluación de riesgo de diferentes enfermedades.
Entre los trabajos que se están realizando actualmente se encuentran estudios genéticos de hemocromatosis hereditaria, trombofilias, marcador genético de celiaquía, análisis antígenos de histocompatibilidad y análisis de traslocaciones cromosómicas, relacionadas con enfermedades oncohematológicas, o fibrosis quística.
La Unidad está dotada con los más modernos equipos y cuenta con personal especializado, en concreto con las profesionales Esther Ocaña, Rosario Aguilar y la responsable del Servicio de Análisis Clínicos, Manuela Gassó.
"La aplicación de estas técnicas de Biología Molecular permitirá la aplicación de tratamientos personalizados en procesos tumorales, que hace algunos años era imposible de imaginar, y la realización de nuevas pruebas a los recién nacidos para la detección precoz de enfermedades raras, como la fibrosis quística, siendo esta una de las líneas prioritarias de la Consejería de Salud", destacó Gassó, que incidió en que, además, la unidad propiciará la solicitud de proyectos que requieran métodos de Biología Molecular.
Hasta el momento, la realización de algunas pruebas genéticas especiales se estaban realizando en laboratorios externos al centro.
Con la implantación de esta Unidad, se ha conseguido realizar estas pruebas en el Complejo Hospitalario de Jaén, permitiendo ofrecer este servicio al resto de hospitales de la provincia.
miércoles, 25 de agosto de 2010
El Clinic y Sant Joan de Déu crean una unidad de adultos con enfermedades raras
http://www.europapress.es/sociedad/salud/noticia-cataluna-clinic-sant-joan-deu-crean-unidad-adultos-enfermedades-raras-20100608124604.html
El Clínic y Sant Joan de Déu crean una unidad de adultos con enfermedades raras
Directorio:Hospital Universitario,enfermedades raras,Investigación Biomédica,
Hospital Clínic
BARCELONA, 8 Jun. (EUROPA PRESS) -
El Hospital Clínic de Barcelona y el Hospital de Sant Joan de Déu en Esplugues de Llobregat (Barcelona) podrán pondrán en marcha una unidad para el diagnóstico y seguimiento de adultos afectados por enfermedades raras, en el marco del Centro de Investigación Biomédica en Red de Enfermedades Raras (Ciberer).
En un comunicado, el Clínic explica que cada vez se diagnostican más casos de enfermedades metabólicas hereditarias y otras patologías minoritarias, al tiempo que los afectados llegan a la edad adulta por la mejora en los tratamientos.
Ello exige equipos multidisciplinares que puedan hacer frente a las complicaciones de tipo neurológico, obstétrico, nutricional o dietético.
El Clínic acoge este martes una jornada sobre Enfermedades Metabólicas Hereditarias en Adultos para abordar los retos en diagnóstico, terapia y seguimiento de los afectados.
Participarán diversos grupos asistenciales implicados en la puesta en marcha de la unidad, así como representantes del Hospital Pitie-Salpêtrière y la Universidad Pierre et Marie Curie de París, uno de los pocos referentes mundiales en la materia.
El Clínic y Sant Joan de Déu crean una unidad de adultos con enfermedades raras
Directorio:Hospital Universitario,enfermedades raras,Investigación Biomédica,
Hospital Clínic
BARCELONA, 8 Jun. (EUROPA PRESS) -
El Hospital Clínic de Barcelona y el Hospital de Sant Joan de Déu en Esplugues de Llobregat (Barcelona) podrán pondrán en marcha una unidad para el diagnóstico y seguimiento de adultos afectados por enfermedades raras, en el marco del Centro de Investigación Biomédica en Red de Enfermedades Raras (Ciberer).
En un comunicado, el Clínic explica que cada vez se diagnostican más casos de enfermedades metabólicas hereditarias y otras patologías minoritarias, al tiempo que los afectados llegan a la edad adulta por la mejora en los tratamientos.
Ello exige equipos multidisciplinares que puedan hacer frente a las complicaciones de tipo neurológico, obstétrico, nutricional o dietético.
El Clínic acoge este martes una jornada sobre Enfermedades Metabólicas Hereditarias en Adultos para abordar los retos en diagnóstico, terapia y seguimiento de los afectados.
Participarán diversos grupos asistenciales implicados en la puesta en marcha de la unidad, así como representantes del Hospital Pitie-Salpêtrière y la Universidad Pierre et Marie Curie de París, uno de los pocos referentes mundiales en la materia.
SEMFYC: Protocolo On Line para pacientes con Enfermedades Raras
http://www.imserso.es/creer_01/documentacion/boletindigitalcreer/ano_2010/julioagosto/monograficomes/index.htm
Protocolo online para la atención de los pacientes con Enfermedades Raras
La semFYC ha elaborado un protocolo online de atención a pacientes con enfermedades raras en la consulta de atención primaria, en colaboración con el Instituto de Investigación de Enfermedades Raras (IIER), perteneciente al Instituto de Salud Carlos III (ISCIII), la Federación Española de Enfermedades Raras (Feder) y el Centro de Referencia Estatal de Atención a Personas con Enfermedades Raras y sus Familias de Burgos (Creer).
CADA MÉDICO DE FAMILIA ATIENDE UNA MEDIA DE 15 PACIENTES AL AÑO CON UNA ENFERMEDAD RARA
3 millones de españoles están afectados por una de estas patologías, que se caracterizan por:
1.- su cronicidad,
2.- baja prevalencia,
3.- elevada morbilidad y mortalidad precoz así como,
4.- en muchos casos, necesidades especiales.
Objetivos:
a.- Mejorar el diagnóstico y la información,
b.- así como la coordinación entre los diferentes especialistas y
c.- facilitar datos epidemiológicos que justifiquen la necesidad de investigar más en estas enfermedades son algunos de los objetivos de este protocolo
El médico de familia debe informar y asesorar al paciente, así como animarle a que entre a formar parte del Registro Epidemiológico del Programa de Investigación en Enfermedades Raras (REpIER), dirigido desde el Instituto de Investigación de Enfermedades Raras.
Madrid, X de junio de 2010 -
Entre 10 y 15 pacientes que pasan al año por la consulta del médico de familia están afectados por una Enfermedad Rara (ER).
Un conjunto de patologías crónicas muy diversas que se caracterizan por su baja prevalencia (menos de 5 casos por cada 10.000 habitantes) elevada morbilidad y mortalidad precoz, lo que determina en muchos casos unas necesidades especiales.
En este contexto, la Sociedad Española de Medicina de Familia y Comunitaria (semFYC), que representa a más de 20.000 profesionales de Atención Primaria de toda España, ha elaborado un protocolo onlinepara la atención de los pacientes con ER en la consulta de Atención Primaria, contando con la colaboración del:
a.- Instituto de Investigación de Enfermedades Raras (IIER), dependiente del Instituto de Salud Carlos III,
b.- de la Federación Española de Enfermedades Raras (Feder) y
c.- del Centro de Referencia Estatal de Atención a Personas con Enfermedades Raras y sus Familias de Burgos (Creer).
Como señala el coordinador del Grupo de Genética Clínica y Enfermedades Raras de semFYC, el Dr. Miguel García Ribes, :
“los objetivos del protocolo son mejorar el diagnóstico y la información que existe en torno a estas patologías, así como la coordinación entre los diferentes especialistas y facilitar datos epidemiológicos que justifiquen la puesta en marcha de acciones en el ámbito de la planificación sanitaria y fomenten la investigación de las ER.
Su baja prevalencia ha condicionado a que existan pocos estudios y los tratamientos en la mayoría de los casos son inexistentes.
Aunque es todavía pronto para asegurar que estas personas tienen cubiertas sus necesidades específicas socio-sanitarias, las autoridades tanto sanitarias como del ámbito social ya empiezan a tomar conciencia de las dimensiones de este problema, que en nuestro país afecta a cerca de tres millones de personas”.
Falta de coordinación entre profesionales
Una de las principales demandas de estos pacientes es la falta de coordinación entre los profesionales.
En este sentido, el protocolo DICE-APER busca mejorar la colaboración asistencial, estableciendo los lazos oportunos con el servicio médico especialista al que ha sido derivado cada paciente.
“En este protocolo”, explica el Doctor García Ribes, “se incluye el envío de una carta, que el médico de familia puede hacer llegar al especialista que lleva cada caso vía correo electrónico o correo postal, en la cual se informará al médico especialista acerca del interés y disposición para que exista una coordinada estrecha entre la atención especializada la correspondiente atención en la consulta de primaria y facilitar sus datos de contacto para poder mantener un intercambio fluido de información sobre la evolución clínica de esa persona”.
Información y diagnóstico
El médico de familia debe confirmar que el diagnóstico que trae el paciente es el de una ER y siempre debe quedar reflejado en su historia clínica.
Asimismo, este profesional juega un papel clave en la educación y asesoría del paciente con una ER.
“Es importante que expliquemos a nuestros pacientes que existe un registro de pacientes de ER y del interés que pertenecer al mismo tiene para la investigación de su enfermedad, por lo que se le invita a formar parte del mismo. El crecimiento de esta base de datos es clave para conocer las dimensiones del problema. Asimismo, el biobanco de ER es una pieza fundamental en la promoción de la investigación y en los análisis que ayuden al diagnóstico y la búsqueda de biomarcadores pronósticos.
Sin embrago, la baja prevalencia de estas patologías dificulta conseguir muestras biológicas que sirvan para llevar a cabo investigaciones de mayos calado científico.
Es imprescindible mostrar al paciente de la importancia de esta donación de una pequeña muestra de sangre, que se haría coincidir con una de sus extracciones analíticas de rutina”.
En opinión del Dr. García Ribes, este protocolo no pretende crear más sobrecarga de trabajo, sino que lo que persigue es ordenar de forma lógica aquellas tareas que podrían ser asumidas por el médico de familia en la atención a estos pacientes.
“De este modo se cubrirían objetivos básicos que facilitarían el control y la gestión de las ER, a la vez que convertiríamos esta faceta de la AP en un modelo de atención exportable a Europa”, concluye este experto.
• Para más información, Gabinete de Prensa semFYC: 91.787.03.00
Protocolo online para la atención de los pacientes con Enfermedades Raras
La semFYC ha elaborado un protocolo online de atención a pacientes con enfermedades raras en la consulta de atención primaria, en colaboración con el Instituto de Investigación de Enfermedades Raras (IIER), perteneciente al Instituto de Salud Carlos III (ISCIII), la Federación Española de Enfermedades Raras (Feder) y el Centro de Referencia Estatal de Atención a Personas con Enfermedades Raras y sus Familias de Burgos (Creer).
CADA MÉDICO DE FAMILIA ATIENDE UNA MEDIA DE 15 PACIENTES AL AÑO CON UNA ENFERMEDAD RARA
3 millones de españoles están afectados por una de estas patologías, que se caracterizan por:
1.- su cronicidad,
2.- baja prevalencia,
3.- elevada morbilidad y mortalidad precoz así como,
4.- en muchos casos, necesidades especiales.
Objetivos:
a.- Mejorar el diagnóstico y la información,
b.- así como la coordinación entre los diferentes especialistas y
c.- facilitar datos epidemiológicos que justifiquen la necesidad de investigar más en estas enfermedades son algunos de los objetivos de este protocolo
El médico de familia debe informar y asesorar al paciente, así como animarle a que entre a formar parte del Registro Epidemiológico del Programa de Investigación en Enfermedades Raras (REpIER), dirigido desde el Instituto de Investigación de Enfermedades Raras.
Madrid, X de junio de 2010 -
Entre 10 y 15 pacientes que pasan al año por la consulta del médico de familia están afectados por una Enfermedad Rara (ER).
Un conjunto de patologías crónicas muy diversas que se caracterizan por su baja prevalencia (menos de 5 casos por cada 10.000 habitantes) elevada morbilidad y mortalidad precoz, lo que determina en muchos casos unas necesidades especiales.
En este contexto, la Sociedad Española de Medicina de Familia y Comunitaria (semFYC), que representa a más de 20.000 profesionales de Atención Primaria de toda España, ha elaborado un protocolo onlinepara la atención de los pacientes con ER en la consulta de Atención Primaria, contando con la colaboración del:
a.- Instituto de Investigación de Enfermedades Raras (IIER), dependiente del Instituto de Salud Carlos III,
b.- de la Federación Española de Enfermedades Raras (Feder) y
c.- del Centro de Referencia Estatal de Atención a Personas con Enfermedades Raras y sus Familias de Burgos (Creer).
Como señala el coordinador del Grupo de Genética Clínica y Enfermedades Raras de semFYC, el Dr. Miguel García Ribes, :
“los objetivos del protocolo son mejorar el diagnóstico y la información que existe en torno a estas patologías, así como la coordinación entre los diferentes especialistas y facilitar datos epidemiológicos que justifiquen la puesta en marcha de acciones en el ámbito de la planificación sanitaria y fomenten la investigación de las ER.
Su baja prevalencia ha condicionado a que existan pocos estudios y los tratamientos en la mayoría de los casos son inexistentes.
Aunque es todavía pronto para asegurar que estas personas tienen cubiertas sus necesidades específicas socio-sanitarias, las autoridades tanto sanitarias como del ámbito social ya empiezan a tomar conciencia de las dimensiones de este problema, que en nuestro país afecta a cerca de tres millones de personas”.
Falta de coordinación entre profesionales
Una de las principales demandas de estos pacientes es la falta de coordinación entre los profesionales.
En este sentido, el protocolo DICE-APER busca mejorar la colaboración asistencial, estableciendo los lazos oportunos con el servicio médico especialista al que ha sido derivado cada paciente.
“En este protocolo”, explica el Doctor García Ribes, “se incluye el envío de una carta, que el médico de familia puede hacer llegar al especialista que lleva cada caso vía correo electrónico o correo postal, en la cual se informará al médico especialista acerca del interés y disposición para que exista una coordinada estrecha entre la atención especializada la correspondiente atención en la consulta de primaria y facilitar sus datos de contacto para poder mantener un intercambio fluido de información sobre la evolución clínica de esa persona”.
Información y diagnóstico
El médico de familia debe confirmar que el diagnóstico que trae el paciente es el de una ER y siempre debe quedar reflejado en su historia clínica.
Asimismo, este profesional juega un papel clave en la educación y asesoría del paciente con una ER.
“Es importante que expliquemos a nuestros pacientes que existe un registro de pacientes de ER y del interés que pertenecer al mismo tiene para la investigación de su enfermedad, por lo que se le invita a formar parte del mismo. El crecimiento de esta base de datos es clave para conocer las dimensiones del problema. Asimismo, el biobanco de ER es una pieza fundamental en la promoción de la investigación y en los análisis que ayuden al diagnóstico y la búsqueda de biomarcadores pronósticos.
Sin embrago, la baja prevalencia de estas patologías dificulta conseguir muestras biológicas que sirvan para llevar a cabo investigaciones de mayos calado científico.
Es imprescindible mostrar al paciente de la importancia de esta donación de una pequeña muestra de sangre, que se haría coincidir con una de sus extracciones analíticas de rutina”.
En opinión del Dr. García Ribes, este protocolo no pretende crear más sobrecarga de trabajo, sino que lo que persigue es ordenar de forma lógica aquellas tareas que podrían ser asumidas por el médico de familia en la atención a estos pacientes.
“De este modo se cubrirían objetivos básicos que facilitarían el control y la gestión de las ER, a la vez que convertiríamos esta faceta de la AP en un modelo de atención exportable a Europa”, concluye este experto.
• Para más información, Gabinete de Prensa semFYC: 91.787.03.00
jueves, 19 de agosto de 2010
Exostosis tibial: Osteocondroma solitario
http://www.pap.es/FrontOffice/PAP/front/Articulos/Articulo/_IXus5l_LjPoo2J2KDAbNm7islArjMMmA
Exostosis tibial: osteocondroma
Autores:
Parada López R a, Montano Navarro E b, Lafraya Puente AL c, Rodríguez Ortega M d
a Pediatra. CS Pinto y Getafe. Servicio Madrileño de Salud, Área 10. Madrid. España.
b Pediatra. CS Pinto y Getafe. Servicio Madrileño de Salud, Área 10. Madrid. España.
c Pediatra. CS Pinto y Getafe. Servicio Madrileño de Salud, Área 10. Madrid. España.
d Cirugía General y Aparato Digestivo. Hospital Infanta Cristina. Parla, Madrid. España.
Correspondencia: R Parada. Correo electrónico: rebecaparada@hotmail.com
Referencia para citar este artículo:
Parada López R, Montano Navarro E, Lafraya Puente AL, Rodríguez Ortega M.
Exostosis tibial: osteocondroma. Rev Pediatr Aten Primaria. 2010;12:255-261.
Resumen:
El osteocondroma o exostosis cartilaginosa es el tumor óseo más frecuente, representando el 10-15% de la totalidad. Alrededor del 3% de la población lo padece.
Es un tumor propio de individuos jóvenes con ligero predominio en varones.
Puede ser solitario o múltiple, formando parte del síndrome de exostosis múltiple hereditaria.
Suele ser un hallazgo accidental, normalmente asintomático, cuya localización más frecuente es la rodilla, aunque puede aparecer en otras localizaciones.
El diagnóstico se realiza por imagen radiográfica, que suele ser patognomónica, y el tratamiento definitivo consiste en la extirpación quirúrgica.
Introducción
Los osteocondromas son lesiones benignas formadoras de cartílago.
Suponen entre el 20-50% de los tumores benignos y entre el 10-20% de todos los tumores primarios del hueso; alrededor del 3% de la población lo padece1-3.
Son de origen cartilaginoso y se conforman tras la separación de un fragmento desde el cartílago epifisario.
Pueden ser solitarios o múltiples, y pueden aparecer espontáneamente o tras un traumatismo. Han sido descritos en prácticamente todos los huesos del esqueleto, pero tienen predilección por la metáfisis de huesos largos.
En este artículo planteamos el caso de un paciente de 13 años de edad que acude a nuestra consulta por presentar una excrecencia a nivel tibial secundaria a este tumor de origen óseo confirmado mediante radiodiagnóstico.
Caso clínico
Varón de 13 años, sin antecedentes personales de interés, que acude a la consulta del centro de salud porque desde hace unos meses ha notado una lesión en la pierna izquierda que no ocasiona ninguna sintomatología y no ha variado de tamaño.
Refiere un posible traumatismo sobre dicha zona 2 años antes tras una caída con patines.
Presenta un buen estado general y se encuentra asintomático.
Se palpa prominencia de aproximadamente 1,5-2 cm de consistencia ósea alrededor de 3-4 cm por encima del maléolo interno izquierdo.
No se objetiva inflamación local, hematoma ni dolor a la palpación.
Tampoco existen alteraciones neurovasculares distales ni impotencia funcional.
Se solicita una radiografía simple de la pierna afecta, anteroposterior y lateral, donde se visualiza una lesión de carácter óseo.
El informe radiológico da el diagnóstico:
“La radiografía anteroposterior y lateral del tobillo muestra un relieve óseo bien delimitado, a expensas de la cortical, sin aparente afectación de partes blandas, en cara medial de tercio distal de la tibia izquierda compatible con osteocondroma”.
Dado que se trata de una masa asintomática y se ha mantenido estable conservando su tamaño, se decide seguir la evolución en la consulta y ante posibles cambios realizar prueba de imagen y/o citarlo en consultas de traumatología para revisión.
Discusión
Nos encontramos ante un caso de osteocondroma, exostosis ósea recubierta de cartílago que se ubica en la superficie externa del hueso.
Es un tumor propio de individuos muy jóvenes; de hecho, el 70% de las lesiones osteocondrales se encuentra en las 2 primeras décadas de la vida2,4.
Tiene un ligero predominio en varones, con una proporción de 3/12.
Los osteocondromas se forman tras la separación de un fragmento desde el cartílago epifisario, que se hernia a través del periostio que envuelve el platillo de crecimiento.
El desarrollo posterior de este fragmento cartilaginoso y su osificación endocondral dará lugar a la exostosis recubierta de cartílago que se proyecta hacia la superficie ósea.
El cartílago extruido sigue el proceso normal de osificación, tiene un espesor menor de 1 cm y es histológicamente normal5.
Los osteocondromas pueden ser solitarios o múltiples1,4, siendo estos últimos constituyentes de la exostosis hereditaria múltiple1,3,6.
En nuestro paciente sólo se encontró una lesión y no había habido ninguna otra persona en su familia con un cuadro similar.
Algunos adolescentes refieren un antecedente de traumatismo o ejercicio vigoroso previo a la aparición del mismo9.
La exostosis puede desarrollarse en cualquier hueso del esqueleto, mostrando preferencia por las zonas metafisarias vecinas a los cartílagos más fértiles de los huesos largos: distal del fémur, proximal de la tibia, distal del radio, etc. La ubicación más frecuente es la rodilla2,4-6.
En ocasiones se desarrollan en huesos de la pelvis, escápula y costillas, tratándose de masas sésiles de pedículo corto. Es raro que afecten a los huesos cortos de manos y pies.
La lesión que comienza precozmente causa trastornos de crecimiento, con acortamiento y deformidad de la extremidad afectada.
Sin embargo, habitualmente es asintomática y se diagnostica como un hallazgo casual1, como el caso que nos ocupa.
Las deformidades esqueléticas y estéticas causadas por los osteocondromas subyacentes son la forma característica de presentación.
Igualmente, pueden producir compresión extrínseca sobre las estructuras óseas, articulares, nerviosas, musculares y ligamentosas vecinas.
También pueden producir compresión de vísceras, como ocurre en los derrames pleurales causados por los osteocondromas costales.
En las zonas de fricción ósea se suelen desarrollar bolsas sinoviales.
Las fracturas son infrecuentes y se producen principalmente en la región de la rodilla2,4,5.
Finalmente, lo más importante a tener en cuenta es que si un osteocondroma duele, además hay que descartar su posible transformación maligna (condrosarcoma)2,3,7.
El riesgo de transformación sarcomatosa en la exostosis solitaria es aproximadamente del 1%, pero en la forma múltiple hereditaria el riesgo se acerca al 10% debido a las numerosas lesiones3. Los datos que nos inducen a pensar que existe una transformación maligna son:
1) la lesión se hace sintomática de repente (duele) o empieza a crecer rápidamente;
2) la cubierta cartilaginosa es más gruesa de 1 cm en un adulto (en el niño puede ser de 2-3 cm de espesor) o con un diámetro máximo mayor de 5 cm;
3) el aumento súbito o marcado en la captación en la gammagrafía ósea en un adulto (en la madurez esquelética lo normal es que esté latente); y
4) la confirmación por la TC (tomografía computadorizada) o RM (resonancia magnética) de una masa de tejidos blandos o desplazamiento de un paquete neurovascular mayor4.
Normalmente los grandes osteocondromas suelen producir desplazamiento de los vasos vecinos. Las complicaciones vasculares de los osteocondromas incluyen: pseudoaneurismas, oclusiones arteriales y venosas y fístulas arteriovenosas.
El 90% de las complicaciones vasculares afectan directamente a las arterias, siendo los pseudoaneurismas la patología más frecuente (63,9%)3-5.
En cuanto al diagnóstico, la prueba de imagen más usada es la radiografía simple.
La apariencia radiográfica de una exostosis es la de una lesión aplanada, sésil o pediculada3-4 (imagen radiológica patognomónica).
Actualmente, la ecografía es el método más accesible en nuestro medio.
Es útil en la valoración del casquete cartilaginoso y además tiene la ventaja de que en los casos de existencia de pseudoaneurisma, muestra con facilidad la presencia de una masa compleja con flujo en su interior y dependiente de una gran arteria.
Otros métodos de imagen son:
la TC (define estructuras óseas anómalas y su relación con complicaciones vasculares mediante la inyección de contraste endovenoso y es de gran utilidad en el estudio del osteocondroma que se origina en hombro, pelvis, columna vertebral y escápula) y la RM, prueba de elección ante la sospecha de malignización. Permite detectar la continuidad entre el hueso medular y cortical del tumor y el hueso afecto. Además el casquete cartilaginoso es hiperintenso en secuencia T21-2.
Cuando en la consulta de Atención Primaria nos encontramos un caso como éste, debemos llevar a cabo un diagnóstico diferencial, aunque su presentación clínica no suele presentar problemas para el diagnóstico. Debemos diferenciarlo principalmente de la exostosis cartilaginosa múltiple y el osteosarcoma parostal.
La exostosis cartilaginosa múltiple es una enfermedad congénita caracterizada por tumores óseos que se distribuyen por casi todo el esqueleto, descrita ya en 1891 por Bessel-Hagen.
Tiene una herencia autosómica dominante.
Se han identificado tres loci en relación con la enfermedad:
EXT1, en el cromosoma 8q23-q24;
EXT2 en el 11p11-p12 y
EXT3 en el brazo corto del cromosoma 19.
Alrededor de 2/3 de los pacientes tienen antecedentes familiares de la enfermedad.
Esta forma múltiple tiene una prevalencia estimada entre el 1/1.000 y el 1/50.000, dependiendo de la zona geográfica que se estudie7.
Se debe buscar esta entidad en pacientes con estatura corta, achatamiento de radio y deformidad angular de los miembros inferiores, signos que no están presentes en nuestro paciente.
El osteosarcoma parostal puede presentarse como una exostosis sintomática que aumenta de tamaño en los adultos.
Está compuesto de tejidos fibro-óseos densos que no están presentes en el osteocondroma.
Con respecto al tratamiento del osteocondroma, existen dos opciones;
a.- por un lado el conservador, en aquellos casos que resultan asintomáticos y se mantienen estables y,
b.-por otro, el quirúrgico, indicado en las formas sintomáticas de la infancia y el crecimiento continuado del osteocondroma después de la madurez del esqueleto (sospecha de malignización)4.
El tratamiento definitivo consiste en una resección simple o ampliada.
Si es pediculado se hace la resección simple hasta la base.
Cuando es sésil se hace una resección ampliada sacando una zona de cortical y completando con curetaje 6, extirpando siempre la exostosis, su cobertura cartilaginosa y el periostio, ya que los restos de éste y del pericondrio pueden dar lugar a una recidiva tumoral.
Con estos procedimientos normalmente se logra la curación 2.
El principal riesgo de la intervención es romper la cortical y la transformación de la lesión en fractura 2,4,7.
Se puede hacer resección quirúrgica de forma profiláctica si se encuentran en vecindad de un vaso, si impiden el movimiento articular normal, en los casos de fracturas o si existe sospecha fundada de transformación maligna2.
El pronóstico generalmente es favorable en los niños, pues cesan con el crecimiento y la madurez ósea.
Alrededor de un 1,8% de los osteocondromas recidivan debido al potencial de crecimiento.
Esta recurrencia consiste en un nuevo osteocondroma, aunque en ocasiones se trata de un condrosarcoma de bajo grado1,2.
El seguimiento de estos pacientes varía de individuo a individuo, dependiendo de la extensión de la enfermedad, el tamaño y la localización del tumor, la respuesta al tratamiento, la edad, el estado general del niño y los avances en el tratamiento.
Las terapias biológicas podrán ser posibles en el futuro1,2.
Bibliografía
1.Dickey IE. Solitary Osteochondroma. E-Medicine & Medscape [Internet] [actualizado el 15/09/2009; consultado el 14/02/2010]. Disponible en http://emedicine.medscape.com/article/1256477-overview
2.Khan AN. Osteochondroma and Osteochondromatosis. E-Medicine & Medscape [Internet] [actualizado el 19/10/2009; consultado el 14/02/2010].
Disponible en http://emedicine.medscape.com/article/392546-overview
3.Staals EL, Bacchini P, Mercuri M, Bertoni F. Dedifferentiated chondrosarcomas arising in preexisting osteochondromas. J Bone Joint Surg Am. 2007;89:987.
4.Florez B, Mönckeberg J, Castillo G, Beguiristain J. Solitary ostechondroma long-term follow-up. J. Pediatr Orthop B. 2008;17(2):91-4.
5.Mavrogenis AF, Papagelopoulos PJ, Soucacos PN. Skeletal ostechondromas revisited. Orthopedics. 2008;31(10).
6.Sepúlveda M. Tumores formadores de cartílago: clínica y tratamiento. Medwave [Internet] [actualizado en 11/2003; consultado el 14/02/ 2010]. Disponible en www.medwave.cl/cursos/Tumores/noviembre2003/3.act.
7.Murphey MD, Chol JJ, Kransdorf MJ, Flemming DJ, Gannon FH. Imaging of osteochondroma: Variants and complications with radiologic pathologic correlation.
Radiographics. 2000;20:1407-34.
8.Children´s Hospital Boston. Ostechondroma (exostosis). [Internet] [consultado el 14/02/ 2010]. Disponible en www.childrenshospital.org/az/Site1079/mainpageS1079P0.html
9.Ortega R, Fernández ME, Gómez I. Masa poplítea asociada a osteocondroma. An Pediatr (Barc). 2005;63(2):185-6.
Exostosis tibial: osteocondroma
Autores:
Parada López R a, Montano Navarro E b, Lafraya Puente AL c, Rodríguez Ortega M d
a Pediatra. CS Pinto y Getafe. Servicio Madrileño de Salud, Área 10. Madrid. España.
b Pediatra. CS Pinto y Getafe. Servicio Madrileño de Salud, Área 10. Madrid. España.
c Pediatra. CS Pinto y Getafe. Servicio Madrileño de Salud, Área 10. Madrid. España.
d Cirugía General y Aparato Digestivo. Hospital Infanta Cristina. Parla, Madrid. España.
Correspondencia: R Parada. Correo electrónico: rebecaparada@hotmail.com
Referencia para citar este artículo:
Parada López R, Montano Navarro E, Lafraya Puente AL, Rodríguez Ortega M.
Exostosis tibial: osteocondroma. Rev Pediatr Aten Primaria. 2010;12:255-261.
Resumen:
El osteocondroma o exostosis cartilaginosa es el tumor óseo más frecuente, representando el 10-15% de la totalidad. Alrededor del 3% de la población lo padece.
Es un tumor propio de individuos jóvenes con ligero predominio en varones.
Puede ser solitario o múltiple, formando parte del síndrome de exostosis múltiple hereditaria.
Suele ser un hallazgo accidental, normalmente asintomático, cuya localización más frecuente es la rodilla, aunque puede aparecer en otras localizaciones.
El diagnóstico se realiza por imagen radiográfica, que suele ser patognomónica, y el tratamiento definitivo consiste en la extirpación quirúrgica.
Introducción
Los osteocondromas son lesiones benignas formadoras de cartílago.
Suponen entre el 20-50% de los tumores benignos y entre el 10-20% de todos los tumores primarios del hueso; alrededor del 3% de la población lo padece1-3.
Son de origen cartilaginoso y se conforman tras la separación de un fragmento desde el cartílago epifisario.
Pueden ser solitarios o múltiples, y pueden aparecer espontáneamente o tras un traumatismo. Han sido descritos en prácticamente todos los huesos del esqueleto, pero tienen predilección por la metáfisis de huesos largos.
En este artículo planteamos el caso de un paciente de 13 años de edad que acude a nuestra consulta por presentar una excrecencia a nivel tibial secundaria a este tumor de origen óseo confirmado mediante radiodiagnóstico.
Caso clínico
Varón de 13 años, sin antecedentes personales de interés, que acude a la consulta del centro de salud porque desde hace unos meses ha notado una lesión en la pierna izquierda que no ocasiona ninguna sintomatología y no ha variado de tamaño.
Refiere un posible traumatismo sobre dicha zona 2 años antes tras una caída con patines.
Presenta un buen estado general y se encuentra asintomático.
Se palpa prominencia de aproximadamente 1,5-2 cm de consistencia ósea alrededor de 3-4 cm por encima del maléolo interno izquierdo.
No se objetiva inflamación local, hematoma ni dolor a la palpación.
Tampoco existen alteraciones neurovasculares distales ni impotencia funcional.
Se solicita una radiografía simple de la pierna afecta, anteroposterior y lateral, donde se visualiza una lesión de carácter óseo.
El informe radiológico da el diagnóstico:
“La radiografía anteroposterior y lateral del tobillo muestra un relieve óseo bien delimitado, a expensas de la cortical, sin aparente afectación de partes blandas, en cara medial de tercio distal de la tibia izquierda compatible con osteocondroma”.
Dado que se trata de una masa asintomática y se ha mantenido estable conservando su tamaño, se decide seguir la evolución en la consulta y ante posibles cambios realizar prueba de imagen y/o citarlo en consultas de traumatología para revisión.
Discusión
Nos encontramos ante un caso de osteocondroma, exostosis ósea recubierta de cartílago que se ubica en la superficie externa del hueso.
Es un tumor propio de individuos muy jóvenes; de hecho, el 70% de las lesiones osteocondrales se encuentra en las 2 primeras décadas de la vida2,4.
Tiene un ligero predominio en varones, con una proporción de 3/12.
Los osteocondromas se forman tras la separación de un fragmento desde el cartílago epifisario, que se hernia a través del periostio que envuelve el platillo de crecimiento.
El desarrollo posterior de este fragmento cartilaginoso y su osificación endocondral dará lugar a la exostosis recubierta de cartílago que se proyecta hacia la superficie ósea.
El cartílago extruido sigue el proceso normal de osificación, tiene un espesor menor de 1 cm y es histológicamente normal5.
Los osteocondromas pueden ser solitarios o múltiples1,4, siendo estos últimos constituyentes de la exostosis hereditaria múltiple1,3,6.
En nuestro paciente sólo se encontró una lesión y no había habido ninguna otra persona en su familia con un cuadro similar.
Algunos adolescentes refieren un antecedente de traumatismo o ejercicio vigoroso previo a la aparición del mismo9.
La exostosis puede desarrollarse en cualquier hueso del esqueleto, mostrando preferencia por las zonas metafisarias vecinas a los cartílagos más fértiles de los huesos largos: distal del fémur, proximal de la tibia, distal del radio, etc. La ubicación más frecuente es la rodilla2,4-6.
En ocasiones se desarrollan en huesos de la pelvis, escápula y costillas, tratándose de masas sésiles de pedículo corto. Es raro que afecten a los huesos cortos de manos y pies.
La lesión que comienza precozmente causa trastornos de crecimiento, con acortamiento y deformidad de la extremidad afectada.
Sin embargo, habitualmente es asintomática y se diagnostica como un hallazgo casual1, como el caso que nos ocupa.
Las deformidades esqueléticas y estéticas causadas por los osteocondromas subyacentes son la forma característica de presentación.
Igualmente, pueden producir compresión extrínseca sobre las estructuras óseas, articulares, nerviosas, musculares y ligamentosas vecinas.
También pueden producir compresión de vísceras, como ocurre en los derrames pleurales causados por los osteocondromas costales.
En las zonas de fricción ósea se suelen desarrollar bolsas sinoviales.
Las fracturas son infrecuentes y se producen principalmente en la región de la rodilla2,4,5.
Finalmente, lo más importante a tener en cuenta es que si un osteocondroma duele, además hay que descartar su posible transformación maligna (condrosarcoma)2,3,7.
El riesgo de transformación sarcomatosa en la exostosis solitaria es aproximadamente del 1%, pero en la forma múltiple hereditaria el riesgo se acerca al 10% debido a las numerosas lesiones3. Los datos que nos inducen a pensar que existe una transformación maligna son:
1) la lesión se hace sintomática de repente (duele) o empieza a crecer rápidamente;
2) la cubierta cartilaginosa es más gruesa de 1 cm en un adulto (en el niño puede ser de 2-3 cm de espesor) o con un diámetro máximo mayor de 5 cm;
3) el aumento súbito o marcado en la captación en la gammagrafía ósea en un adulto (en la madurez esquelética lo normal es que esté latente); y
4) la confirmación por la TC (tomografía computadorizada) o RM (resonancia magnética) de una masa de tejidos blandos o desplazamiento de un paquete neurovascular mayor4.
Normalmente los grandes osteocondromas suelen producir desplazamiento de los vasos vecinos. Las complicaciones vasculares de los osteocondromas incluyen: pseudoaneurismas, oclusiones arteriales y venosas y fístulas arteriovenosas.
El 90% de las complicaciones vasculares afectan directamente a las arterias, siendo los pseudoaneurismas la patología más frecuente (63,9%)3-5.
En cuanto al diagnóstico, la prueba de imagen más usada es la radiografía simple.
La apariencia radiográfica de una exostosis es la de una lesión aplanada, sésil o pediculada3-4 (imagen radiológica patognomónica).
Actualmente, la ecografía es el método más accesible en nuestro medio.
Es útil en la valoración del casquete cartilaginoso y además tiene la ventaja de que en los casos de existencia de pseudoaneurisma, muestra con facilidad la presencia de una masa compleja con flujo en su interior y dependiente de una gran arteria.
Otros métodos de imagen son:
la TC (define estructuras óseas anómalas y su relación con complicaciones vasculares mediante la inyección de contraste endovenoso y es de gran utilidad en el estudio del osteocondroma que se origina en hombro, pelvis, columna vertebral y escápula) y la RM, prueba de elección ante la sospecha de malignización. Permite detectar la continuidad entre el hueso medular y cortical del tumor y el hueso afecto. Además el casquete cartilaginoso es hiperintenso en secuencia T21-2.
Cuando en la consulta de Atención Primaria nos encontramos un caso como éste, debemos llevar a cabo un diagnóstico diferencial, aunque su presentación clínica no suele presentar problemas para el diagnóstico. Debemos diferenciarlo principalmente de la exostosis cartilaginosa múltiple y el osteosarcoma parostal.
La exostosis cartilaginosa múltiple es una enfermedad congénita caracterizada por tumores óseos que se distribuyen por casi todo el esqueleto, descrita ya en 1891 por Bessel-Hagen.
Tiene una herencia autosómica dominante.
Se han identificado tres loci en relación con la enfermedad:
EXT1, en el cromosoma 8q23-q24;
EXT2 en el 11p11-p12 y
EXT3 en el brazo corto del cromosoma 19.
Alrededor de 2/3 de los pacientes tienen antecedentes familiares de la enfermedad.
Esta forma múltiple tiene una prevalencia estimada entre el 1/1.000 y el 1/50.000, dependiendo de la zona geográfica que se estudie7.
Se debe buscar esta entidad en pacientes con estatura corta, achatamiento de radio y deformidad angular de los miembros inferiores, signos que no están presentes en nuestro paciente.
El osteosarcoma parostal puede presentarse como una exostosis sintomática que aumenta de tamaño en los adultos.
Está compuesto de tejidos fibro-óseos densos que no están presentes en el osteocondroma.
Con respecto al tratamiento del osteocondroma, existen dos opciones;
a.- por un lado el conservador, en aquellos casos que resultan asintomáticos y se mantienen estables y,
b.-por otro, el quirúrgico, indicado en las formas sintomáticas de la infancia y el crecimiento continuado del osteocondroma después de la madurez del esqueleto (sospecha de malignización)4.
El tratamiento definitivo consiste en una resección simple o ampliada.
Si es pediculado se hace la resección simple hasta la base.
Cuando es sésil se hace una resección ampliada sacando una zona de cortical y completando con curetaje 6, extirpando siempre la exostosis, su cobertura cartilaginosa y el periostio, ya que los restos de éste y del pericondrio pueden dar lugar a una recidiva tumoral.
Con estos procedimientos normalmente se logra la curación 2.
El principal riesgo de la intervención es romper la cortical y la transformación de la lesión en fractura 2,4,7.
Se puede hacer resección quirúrgica de forma profiláctica si se encuentran en vecindad de un vaso, si impiden el movimiento articular normal, en los casos de fracturas o si existe sospecha fundada de transformación maligna2.
El pronóstico generalmente es favorable en los niños, pues cesan con el crecimiento y la madurez ósea.
Alrededor de un 1,8% de los osteocondromas recidivan debido al potencial de crecimiento.
Esta recurrencia consiste en un nuevo osteocondroma, aunque en ocasiones se trata de un condrosarcoma de bajo grado1,2.
El seguimiento de estos pacientes varía de individuo a individuo, dependiendo de la extensión de la enfermedad, el tamaño y la localización del tumor, la respuesta al tratamiento, la edad, el estado general del niño y los avances en el tratamiento.
Las terapias biológicas podrán ser posibles en el futuro1,2.
Bibliografía
1.Dickey IE. Solitary Osteochondroma. E-Medicine & Medscape [Internet] [actualizado el 15/09/2009; consultado el 14/02/2010]. Disponible en http://emedicine.medscape.com/article/1256477-overview
2.Khan AN. Osteochondroma and Osteochondromatosis. E-Medicine & Medscape [Internet] [actualizado el 19/10/2009; consultado el 14/02/2010].
Disponible en http://emedicine.medscape.com/article/392546-overview
3.Staals EL, Bacchini P, Mercuri M, Bertoni F. Dedifferentiated chondrosarcomas arising in preexisting osteochondromas. J Bone Joint Surg Am. 2007;89:987.
4.Florez B, Mönckeberg J, Castillo G, Beguiristain J. Solitary ostechondroma long-term follow-up. J. Pediatr Orthop B. 2008;17(2):91-4.
5.Mavrogenis AF, Papagelopoulos PJ, Soucacos PN. Skeletal ostechondromas revisited. Orthopedics. 2008;31(10).
6.Sepúlveda M. Tumores formadores de cartílago: clínica y tratamiento. Medwave [Internet] [actualizado en 11/2003; consultado el 14/02/ 2010]. Disponible en www.medwave.cl/cursos/Tumores/noviembre2003/3.act.
7.Murphey MD, Chol JJ, Kransdorf MJ, Flemming DJ, Gannon FH. Imaging of osteochondroma: Variants and complications with radiologic pathologic correlation.
Radiographics. 2000;20:1407-34.
8.Children´s Hospital Boston. Ostechondroma (exostosis). [Internet] [consultado el 14/02/ 2010]. Disponible en www.childrenshospital.org/az/Site1079/mainpageS1079P0.html
9.Ortega R, Fernández ME, Gómez I. Masa poplítea asociada a osteocondroma. An Pediatr (Barc). 2005;63(2):185-6.
martes, 17 de agosto de 2010
¿Que son los Osteocondromas en el hombro?
http://www.thehouseofblogs.com/articulo/revistas_gratuitas__dolor_de_hombro_en_paciente-121720.html
Revistas gratuitas : Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico
Fuente: La Medicina Natural y Remedios Naturales.16 del 8 de 2010
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico
http://www.portalesmedicos.com/publicaciones/articles/2089/1/Dolor-de-hombro-en-paciente-con-exostosis-multiple-A-proposito-de-un-caso-clinico.html
Dolor de hombro en paciente con exóstosis múltiple. A propósito de un caso clínico
Mesa Rivero M. E. (MIR),
Rodríguez de la Cueva J. M. (FEA),
González Herranz J. (FEA),
Angulo Gutiérrez J. (FEA).
Servicio de Cirugía Ortopédica y Traumatología. Hospital Universitario de Valme. Sevilla (España).
Dolor de hombro en paciente con exóstosis múltiple. A propósito de un caso.
1. Resumen:
Objetivo: La exóstosis hereditaria múltiple es una enfermedad autosómica dominante caracterizada por la aparición de osteocondromas, de predominio metafisario de los huesos largos (1) y que presentan un recubrimiento cartilaginoso benigno.
Los osteocondromas crecen y se desarrollan en la primera década de la vida. Suelen ser asintomáticos, aunque pueden ocasionar dolor local y compresión de estructuras vasculonerviosas adyacentes o degeneración maligna hacia condrosarcoma, en un 0.5-5% de los casos (2).
Material y método: Presentamos el caso de un paciente de 12 años de edad, afectado de exóstosis múltiple, que acude a nuestras consultas con dolor en hombro izquierdo.
Se realizó tratamiento mediante exéresis de un pedículo tumoral, desapareciendo la clínica y evolucionando favorablemente.
Conclusión: En el estudio anatomopatológico se diagnóstico de osteocondromas múltiples. Consideramos la importancia del caso debido al potencial de malignización de estas lesiones así como la necesidad de seguimiento genésico de los pacientes.
2. Abstract:
Hereditary multiple exostoses is an autosomal dominant inheritance pattern, characteriszed by benign cartilage-capped bone tumours (osteochondromas) that grow outward from the metaphyses of the long bones.
Osteochondromas develop and increase in size in the first decade of life.
The majority are asymptomatic, they may even cause pain and impingement on adjacents nerves and vessels or malignant transformation of osteochondroma towards chondrosarcoma (0.5-5%).
We present a case of a 12 year-old patient with multiple exostoses and left shoulder pain.
We resected a humeral tumour.
The symptoms disappeared, with a good clinic evolution.
Anatomicopathologists carried out a diagnosis of multiple osteochondromas.
We consider the relevance of this case because of the malignant degeneration.
Patients should be well instructed and followed-up for detection of malignancy.
Key words: osteochondromas, multiple exostoses, chondrosarcomas.
3. Introducción:
La exóstosis hereditaria múltiple constituye una entidad caracterizada por el desarrollo de 20 ó más tumores de localización metafisaria y que, típicamente, presentan un recubrimiento cartilaginoso (1).
Su prevalencia es del 1 entre 50.000 habitantes, siendo más frecuente en los varones (2).
En su origen está implicada la mutación de los genes EXT1 Y EXT2, presentando un patrón de herencia autosómica dominante (3).
Los osteocondromas crecen y se desarrollan en la primera década de la vida, con un número de localizaciones variables entre los individuos de una misma familia.
Se localizan principalmente en la metáfisis de los huesos largos, sobre todo próximos a la rodilla. La mayoría son asintomáticos aunque, en una menor proporción pueden producir dolor, compresión de estructuras adyacentes, deformidad, problemas funcionales y degeneración maligna hacia condrosarcoma, justificando dicha sintomatología la exéresis tumoral (2).
El diagnóstico se realiza mediante la clínica del paciente, apoyado por la pruebas de imagen (4) y, a ser posible, complementándose con la historia familiar y el estudio genético de la misma.
El tratamiento quirúrgico mediante la exéresis tumoral está indicado cuando aparecen complicaciones derivadas por un excesivo crecimiento ó por la malignización.
Los osteocondromas son lesiones benignas, dependiendo así el pronóstico de la degeneración maligna hacia condrosarcoma (5).
El patrón de transmisión es autosómico dominante, con una penetrancia del 100%; así, en caso de aislarse el gen afectado, existe la posibilidad de realizar el diagnóstico prenatal (2).
4. Material y método:
Paciente varón de 12 años de edad, que acude al Servicio de Urgencias de Traumatología al presentar dolor en hombro izquierdo de inicio súbito y sin traumatismo previo.
No existen antecedentes personales de interés.
Como antecedentes familiares refiere que 2 hermanos de su madre han sido diagnosticados de exostosis múltiple, sin datos de degeneración maligna en ninguno de ellos.
A la exploración presenta una talla baja para su edad, con extremidades relativamente cortas con respecto al tronco, así como prominencias óseas a nivel de las rodillas, muñecas y hombros.
Consideramos como significativo una limitación de la prono-supinación en ambos extremidades superiores. A la palpación refiere dolor en la zona deltoidea de miembro superior izquierdo y limitación de las rotaciones en ambas articulaciones coxo-femorales.
Realizamos un mapa óseo, hallándose lesiones óseas de predominio metafisario en las tibias, fémures y húmeros, compatibles con el diagnostico de osteocondromatosis.
Destaca la presencia de una fractura del pedículo de un osteocondroma en la metáfisis del húmero izquierdo, así como de un ensanchamiento de la metáfisis del húmero derecho debido a la presencia de varios osteocondromas.
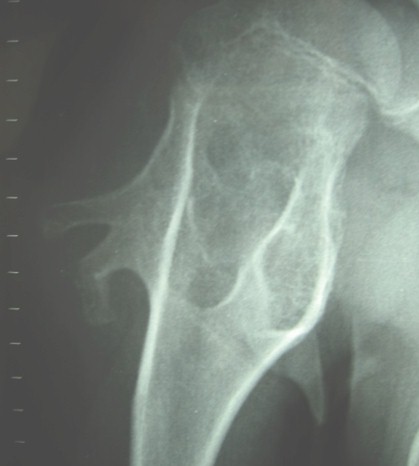
Ampliamos el estudio realizando una TAC de la extremidad proximal de ambos húmeros, donde lo más destacado es la fractura del pedículo óseo antes mencionado en el húmero izquierdo, y la presencia de múltiples osteocondromas en la región proximal de húmero y escápula derecha, llamando especialmente la atención la presencia de un osteocondroma con mayor densidad ósea, en la cara interna de la escápula, que separa a esta de la pared torácica.
En la RMN no se observan, signos inflamatorios, formación de alguna colección, o otras alteraciones en las partes blandas que nos hagan sospechar malignización del proceso.
Realizamos la exéresis transpedicular de 2 osteocondromas en escápula derecha; así como la extirpación, por vía deltopectoral, de 3 tumores en el extremo proximal del húmero izquierdo.
El material obtenido es remitido como material de biopsia para estudio anatomo-patológico.
5. Resultado:
El resultado del estudio anatomopatológico, reveló la presencia de lesiones de tipo osteocartilaginoso, con centro compuesto por material osteoide y recubrimiento superficial de cartílago. Siendo diagnosticado de osteocondromas.
El paciente fue dado de alta con tratamiento analgésicos habitual y, realizando controles periódicos para el seguimiento de su proceso en régimen ambulatorio y evitar una demora en el diagnostico de una posible degeneración maligna.
6. Discusión:
La exóstosis múltiple es una enfermedad que presenta típicamente tumores (osteocondromas) de predominio metafisario y recubiertos por cartílago, apareciendo sobre todo en los huesos largos (1).
La prevalencia es variable, siendo de 1 cada 100.000 individuos en Europa y de 1/50.000 en el estado de Washington (1).
Son más frecuentes en el varón, existiendo una relación de 1.5: 1, entre hombres y mujeres.
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico .2
Los osteocondromas solitarios son 6 veces más frecuentes que la exóstosis múltiple.
El 62% de los pacientes afectados, presentan historia familiar positiva para la enfermedad (3).
En nuestro caso, el paciente presenta un antecedente familiar, 2 tíos varones por vía materna, existiendo la sospecha de afectación familiar en 3 generaciones anteriores.
La historia familiar sugiere un patrón de herencia autosómico dominante, con una penetrancia de casi el 100%.
Parece que en el desarrollo de la enfermedad se ha implicado la mutación del gen EXT, que actúa como un gen supresor tumoral.
Dicho gen actúa como productor de glicosiltranferasas, relacionadas con la síntesis de un recubrimiento celular de heparan sulfato, afectando así la difusión del morfogen Hedgehog (5).
Se ha identificado una mutación cromosómica en tres localizaciones distintas:
EXT 1 (cromosoma 8, locus 8q224.1);
EXT 2 (cromosoma 11, en la región pericéntrica);
EXT 3 (cromosoma 19, locus 19 p),
siendo la mutación del gen EXT 1 la más frecuente cuando existe historia familiar de la enfermedad (1).
El proceso de malignización está representado por una inestabilidad cromosómica, previamente demostrada por un alto porcentaje de pérdida de heterocigosidad, y que implica a varios loci en un amplio rango del DNA.
En el caso presentado no hemos realizado estudio genético.
La proporción de individuos que presentan sintomatología clínica incrementa con la edad, siendo bajo al nacer (5%), aumentado con edad (a los 12 años el 96% ha presentado alguna clínica).
En la edad puberal y, coincidiendo con el cierre del cartílago de crecimiento, es cuando los osteocondromas dejan de crecer (1).
Los síntomas principales de la exóstosis múltiple son:
a.- el dolor,
b.- la limitación funcional de alguna articulación,
c.- es frecuente el acortamiento del cúbito que ocasiona una incurvación del radio (39-60% de los casos),
d.- así como la dismetría de las extremidades (10-50%), y
e.- las deformidades angulares en varo ó valgo de la rodilla (8-33%),
f.- como de los tobillos (2-54%).
g.- Con frecuencia se asocia talla baja ¿rizomélica? (37-44%).
En menor proporción podemos encontrarnos con otras manifestaciones osteoarticulares como bursitis, compresión de estructuras vasculonerviosas, fracturas del pedículo, y dolores difusos generalizados. (3)
La complicación más importante es la degeneración maligna en condrosarcoma, que se presenta entre un 0.5-5% de los casos, y que suele manifestarse como dolor y aumento de la masa tumoral; así como engrosamiento de la capa cartilaginosa que recubre al tumor en más de 1 cm..
La degeneración hacia otras lesiones malignas como fibrosarcoma, fribrohistiocitoma maligno y osteosarcoma, se presenta con menor frecuencia (5).
Los osteocondromas son raros en el esqueleto axial, habiéndose descrito algunos casos de compresión medular a nivel de C1, C2 y C5.
Si sospechamos que un paciente padece de exóstosis múltiple, debemos realizar:
1.- detallado estudio de la historia familiar,
2.- un estudio radiográfico e histológico tumoral y,
3.- cuando sea posible, un estudio genético.
El estudio radiográfico se inicia mediante un mapa óseo, siendo necesario una TAC para poder valorar con exactitud las lesiones y esclarecer el origen de dichas lesiones (2).
Con el TAC podemos realizar un estudio adecuado de la anatomía ósea regional y de las fracturas de las lesiones exofíticas (5).
La RMN con contraste nos permite identificar tanto la morfología del cartílago que recubre al tumor, como de las partes blandas peritumorales, que nos podrá indicar una posible degeneración maligna (5).
En nuestro caso realizamos un mapa óseo al paciente, así como una TAC de la extremidad superior de ambos húmeros debido a los hallazgos previos radiográficos, con la intención de identificar la extensión y crecimiento de los osteocondromas de estas regiones.
Las imágenes de la RNM nos ayudo a descartar la malignización de las lesiones más bizarras.
Debemos realizar un diagnóstico diferencial con:
a.- la Displasia Hemimélica Epifisaria (enfermedad de Trévor) y
b.- la metacondromatosis, enfermedades que no guardan relación con una mutación del gen EXT;
c.- así como diferenciarlo de la enfermedad de Mafucci y Ollier, cuyo patrón de presentación es distinto (3).
El manejo de la exóstosis múltiple, así como su tratamiento, es muy variado.
Cuando dichas lesiones causan dolor, compresión de estructuras adyacentes, deformidad estética ó limitación funcional, está indicado el tratamiento quirúrgico, realizando la exéresis de los osteocondromas que causan estas complicaciones (3).
El tratamiento de la enfermedad del antebrazo es controvertido.
Akita et al. en una serie de 23 pacientes, concluyeron que la osteotomía y el alargamiento de los huesos del antebrazo no era muy beneficiosa.
Así, el procedimiento más adecuado, sería la exéresis del tumor, mejorando la movilidad cuando la lesión afecta al extremo distal del cúbito (3).
Hay que advertir al paciente y sus familiares que debe ser revisado de forma periódica y que en el caso que surja algún cambio en la evolución que solicite una consulta médica de forma inmediata.
Tras la pubertad, los osteocondromas no deben de aumentar de tamaño, así una vez alcanzada la madurez esquelética, se recomienda realizar controles anuales, aunque aún no se ha demostrado la eficacia de dicho seguimiento (3).
En el caso de sufrir la malignización de un osteocondroma, está indicada la resección en bloque, incluyendo la pseudocápsula tumoral y márgenes libres de tumor, ya que así podríamos lograr un buen resultado local y a largo plazo.
A nuestro paciente se le realizó:
1.- exéresis transpedicular del tumor de escápula derecha; y
2.- la exéresis del tumor sésil localizado en metáfisis de húmero izquierdo, cuya rotura era la responsable del dolor que presentaba en hombro izquierdo.
Los osteocondromas son tumores benignos y no afectan la esperanza de vida (3).
El riesgo de malignización hacia osteosarcoma es aproximadamente del 1% y, en caso de producirse, el pronóstico dependerá del grado histológico, siendo:
a.- la supervivencia a los 10 años del 83% para el grado I y
b.- del 29% para el grado III (3).
7. Bibliografía:
1. Gupta PP, Agarwal D. A middle-aged manwith persisting chest opacity and permanent bony swelling. CMAJ 2006 Nov 15 ; 31 (24): E920-4
2. Manish Chadha, MS; Arun Pal Singh, MS. Secondary chondrosarcoma of the cuboid bone in a patient with multiple exostosis. Can J Surg, 2008.Vol. 51 (1).
3. Judith VMG Bovée. Multiple osteochondromas. Orphanet Journal of Rare Diseases 2008, 3:3
4. Carolyn M. Sofka, MD& Gregory R. Saboeiro, MD& Robert Schneider, MD. Multiple Hereditary Exostoses. HSSJ (2005) 1:49–51.
5. J V M G Bovée, R J B Sakkers, M J A Geirnaerdt, A H M Taminiau, P C W Hogendoorn. Intermediate grade osteosarcoma and chondrosarcoma
6. Arising in an osteochondroma. A case report of a patient with hereditary multiple exostoses.J Clin Pathol 2002;55:226–229
Fuente: Portalesmadicos.com
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico
Blogalaxia Tags: medicina medicina alternativa tratamientos naturales remedios caseros remedios para
Webs Recomendadas: remedios y tratamientos naturalesmedicina natural y tratamientostratamientos naturalesTratamientos Medicinales Gratis
Revistas gratuitas : Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico
Fuente: La Medicina Natural y Remedios Naturales.16 del 8 de 2010
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico
http://www.portalesmedicos.com/publicaciones/articles/2089/1/Dolor-de-hombro-en-paciente-con-exostosis-multiple-A-proposito-de-un-caso-clinico.html
Dolor de hombro en paciente con exóstosis múltiple. A propósito de un caso clínico
Mesa Rivero M. E. (MIR),
Rodríguez de la Cueva J. M. (FEA),
González Herranz J. (FEA),
Angulo Gutiérrez J. (FEA).
Servicio de Cirugía Ortopédica y Traumatología. Hospital Universitario de Valme. Sevilla (España).
Dolor de hombro en paciente con exóstosis múltiple. A propósito de un caso.
1. Resumen:
Objetivo: La exóstosis hereditaria múltiple es una enfermedad autosómica dominante caracterizada por la aparición de osteocondromas, de predominio metafisario de los huesos largos (1) y que presentan un recubrimiento cartilaginoso benigno.
Los osteocondromas crecen y se desarrollan en la primera década de la vida. Suelen ser asintomáticos, aunque pueden ocasionar dolor local y compresión de estructuras vasculonerviosas adyacentes o degeneración maligna hacia condrosarcoma, en un 0.5-5% de los casos (2).
Material y método: Presentamos el caso de un paciente de 12 años de edad, afectado de exóstosis múltiple, que acude a nuestras consultas con dolor en hombro izquierdo.
Se realizó tratamiento mediante exéresis de un pedículo tumoral, desapareciendo la clínica y evolucionando favorablemente.
Conclusión: En el estudio anatomopatológico se diagnóstico de osteocondromas múltiples. Consideramos la importancia del caso debido al potencial de malignización de estas lesiones así como la necesidad de seguimiento genésico de los pacientes.
2. Abstract:
Hereditary multiple exostoses is an autosomal dominant inheritance pattern, characteriszed by benign cartilage-capped bone tumours (osteochondromas) that grow outward from the metaphyses of the long bones.
Osteochondromas develop and increase in size in the first decade of life.
The majority are asymptomatic, they may even cause pain and impingement on adjacents nerves and vessels or malignant transformation of osteochondroma towards chondrosarcoma (0.5-5%).
We present a case of a 12 year-old patient with multiple exostoses and left shoulder pain.
We resected a humeral tumour.
The symptoms disappeared, with a good clinic evolution.
Anatomicopathologists carried out a diagnosis of multiple osteochondromas.
We consider the relevance of this case because of the malignant degeneration.
Patients should be well instructed and followed-up for detection of malignancy.
Key words: osteochondromas, multiple exostoses, chondrosarcomas.
3. Introducción:
La exóstosis hereditaria múltiple constituye una entidad caracterizada por el desarrollo de 20 ó más tumores de localización metafisaria y que, típicamente, presentan un recubrimiento cartilaginoso (1).
Su prevalencia es del 1 entre 50.000 habitantes, siendo más frecuente en los varones (2).
En su origen está implicada la mutación de los genes EXT1 Y EXT2, presentando un patrón de herencia autosómica dominante (3).
Los osteocondromas crecen y se desarrollan en la primera década de la vida, con un número de localizaciones variables entre los individuos de una misma familia.
Se localizan principalmente en la metáfisis de los huesos largos, sobre todo próximos a la rodilla. La mayoría son asintomáticos aunque, en una menor proporción pueden producir dolor, compresión de estructuras adyacentes, deformidad, problemas funcionales y degeneración maligna hacia condrosarcoma, justificando dicha sintomatología la exéresis tumoral (2).
El diagnóstico se realiza mediante la clínica del paciente, apoyado por la pruebas de imagen (4) y, a ser posible, complementándose con la historia familiar y el estudio genético de la misma.
El tratamiento quirúrgico mediante la exéresis tumoral está indicado cuando aparecen complicaciones derivadas por un excesivo crecimiento ó por la malignización.
Los osteocondromas son lesiones benignas, dependiendo así el pronóstico de la degeneración maligna hacia condrosarcoma (5).
El patrón de transmisión es autosómico dominante, con una penetrancia del 100%; así, en caso de aislarse el gen afectado, existe la posibilidad de realizar el diagnóstico prenatal (2).
4. Material y método:
Paciente varón de 12 años de edad, que acude al Servicio de Urgencias de Traumatología al presentar dolor en hombro izquierdo de inicio súbito y sin traumatismo previo.
No existen antecedentes personales de interés.
Como antecedentes familiares refiere que 2 hermanos de su madre han sido diagnosticados de exostosis múltiple, sin datos de degeneración maligna en ninguno de ellos.
A la exploración presenta una talla baja para su edad, con extremidades relativamente cortas con respecto al tronco, así como prominencias óseas a nivel de las rodillas, muñecas y hombros.
Consideramos como significativo una limitación de la prono-supinación en ambos extremidades superiores. A la palpación refiere dolor en la zona deltoidea de miembro superior izquierdo y limitación de las rotaciones en ambas articulaciones coxo-femorales.
Realizamos un mapa óseo, hallándose lesiones óseas de predominio metafisario en las tibias, fémures y húmeros, compatibles con el diagnostico de osteocondromatosis.
Destaca la presencia de una fractura del pedículo de un osteocondroma en la metáfisis del húmero izquierdo, así como de un ensanchamiento de la metáfisis del húmero derecho debido a la presencia de varios osteocondromas.
Ampliamos el estudio realizando una TAC de la extremidad proximal de ambos húmeros, donde lo más destacado es la fractura del pedículo óseo antes mencionado en el húmero izquierdo, y la presencia de múltiples osteocondromas en la región proximal de húmero y escápula derecha, llamando especialmente la atención la presencia de un osteocondroma con mayor densidad ósea, en la cara interna de la escápula, que separa a esta de la pared torácica.
En la RMN no se observan, signos inflamatorios, formación de alguna colección, o otras alteraciones en las partes blandas que nos hagan sospechar malignización del proceso.
Realizamos la exéresis transpedicular de 2 osteocondromas en escápula derecha; así como la extirpación, por vía deltopectoral, de 3 tumores en el extremo proximal del húmero izquierdo.
El material obtenido es remitido como material de biopsia para estudio anatomo-patológico.
5. Resultado:
El resultado del estudio anatomopatológico, reveló la presencia de lesiones de tipo osteocartilaginoso, con centro compuesto por material osteoide y recubrimiento superficial de cartílago. Siendo diagnosticado de osteocondromas.
El paciente fue dado de alta con tratamiento analgésicos habitual y, realizando controles periódicos para el seguimiento de su proceso en régimen ambulatorio y evitar una demora en el diagnostico de una posible degeneración maligna.
6. Discusión:
La exóstosis múltiple es una enfermedad que presenta típicamente tumores (osteocondromas) de predominio metafisario y recubiertos por cartílago, apareciendo sobre todo en los huesos largos (1).
La prevalencia es variable, siendo de 1 cada 100.000 individuos en Europa y de 1/50.000 en el estado de Washington (1).
Son más frecuentes en el varón, existiendo una relación de 1.5: 1, entre hombres y mujeres.
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico .2
Los osteocondromas solitarios son 6 veces más frecuentes que la exóstosis múltiple.
El 62% de los pacientes afectados, presentan historia familiar positiva para la enfermedad (3).
En nuestro caso, el paciente presenta un antecedente familiar, 2 tíos varones por vía materna, existiendo la sospecha de afectación familiar en 3 generaciones anteriores.
La historia familiar sugiere un patrón de herencia autosómico dominante, con una penetrancia de casi el 100%.
Parece que en el desarrollo de la enfermedad se ha implicado la mutación del gen EXT, que actúa como un gen supresor tumoral.
Dicho gen actúa como productor de glicosiltranferasas, relacionadas con la síntesis de un recubrimiento celular de heparan sulfato, afectando así la difusión del morfogen Hedgehog (5).
Se ha identificado una mutación cromosómica en tres localizaciones distintas:
EXT 1 (cromosoma 8, locus 8q224.1);
EXT 2 (cromosoma 11, en la región pericéntrica);
EXT 3 (cromosoma 19, locus 19 p),
siendo la mutación del gen EXT 1 la más frecuente cuando existe historia familiar de la enfermedad (1).
El proceso de malignización está representado por una inestabilidad cromosómica, previamente demostrada por un alto porcentaje de pérdida de heterocigosidad, y que implica a varios loci en un amplio rango del DNA.
En el caso presentado no hemos realizado estudio genético.
La proporción de individuos que presentan sintomatología clínica incrementa con la edad, siendo bajo al nacer (5%), aumentado con edad (a los 12 años el 96% ha presentado alguna clínica).
En la edad puberal y, coincidiendo con el cierre del cartílago de crecimiento, es cuando los osteocondromas dejan de crecer (1).
Los síntomas principales de la exóstosis múltiple son:
a.- el dolor,
b.- la limitación funcional de alguna articulación,
c.- es frecuente el acortamiento del cúbito que ocasiona una incurvación del radio (39-60% de los casos),
d.- así como la dismetría de las extremidades (10-50%), y
e.- las deformidades angulares en varo ó valgo de la rodilla (8-33%),
f.- como de los tobillos (2-54%).
g.- Con frecuencia se asocia talla baja ¿rizomélica? (37-44%).
En menor proporción podemos encontrarnos con otras manifestaciones osteoarticulares como bursitis, compresión de estructuras vasculonerviosas, fracturas del pedículo, y dolores difusos generalizados. (3)
La complicación más importante es la degeneración maligna en condrosarcoma, que se presenta entre un 0.5-5% de los casos, y que suele manifestarse como dolor y aumento de la masa tumoral; así como engrosamiento de la capa cartilaginosa que recubre al tumor en más de 1 cm..
La degeneración hacia otras lesiones malignas como fibrosarcoma, fribrohistiocitoma maligno y osteosarcoma, se presenta con menor frecuencia (5).
Los osteocondromas son raros en el esqueleto axial, habiéndose descrito algunos casos de compresión medular a nivel de C1, C2 y C5.
Si sospechamos que un paciente padece de exóstosis múltiple, debemos realizar:
1.- detallado estudio de la historia familiar,
2.- un estudio radiográfico e histológico tumoral y,
3.- cuando sea posible, un estudio genético.
El estudio radiográfico se inicia mediante un mapa óseo, siendo necesario una TAC para poder valorar con exactitud las lesiones y esclarecer el origen de dichas lesiones (2).
Con el TAC podemos realizar un estudio adecuado de la anatomía ósea regional y de las fracturas de las lesiones exofíticas (5).
La RMN con contraste nos permite identificar tanto la morfología del cartílago que recubre al tumor, como de las partes blandas peritumorales, que nos podrá indicar una posible degeneración maligna (5).
En nuestro caso realizamos un mapa óseo al paciente, así como una TAC de la extremidad superior de ambos húmeros debido a los hallazgos previos radiográficos, con la intención de identificar la extensión y crecimiento de los osteocondromas de estas regiones.
Las imágenes de la RNM nos ayudo a descartar la malignización de las lesiones más bizarras.
Debemos realizar un diagnóstico diferencial con:
a.- la Displasia Hemimélica Epifisaria (enfermedad de Trévor) y
b.- la metacondromatosis, enfermedades que no guardan relación con una mutación del gen EXT;
c.- así como diferenciarlo de la enfermedad de Mafucci y Ollier, cuyo patrón de presentación es distinto (3).
El manejo de la exóstosis múltiple, así como su tratamiento, es muy variado.
Cuando dichas lesiones causan dolor, compresión de estructuras adyacentes, deformidad estética ó limitación funcional, está indicado el tratamiento quirúrgico, realizando la exéresis de los osteocondromas que causan estas complicaciones (3).
El tratamiento de la enfermedad del antebrazo es controvertido.
Akita et al. en una serie de 23 pacientes, concluyeron que la osteotomía y el alargamiento de los huesos del antebrazo no era muy beneficiosa.
Así, el procedimiento más adecuado, sería la exéresis del tumor, mejorando la movilidad cuando la lesión afecta al extremo distal del cúbito (3).
Hay que advertir al paciente y sus familiares que debe ser revisado de forma periódica y que en el caso que surja algún cambio en la evolución que solicite una consulta médica de forma inmediata.
Tras la pubertad, los osteocondromas no deben de aumentar de tamaño, así una vez alcanzada la madurez esquelética, se recomienda realizar controles anuales, aunque aún no se ha demostrado la eficacia de dicho seguimiento (3).
En el caso de sufrir la malignización de un osteocondroma, está indicada la resección en bloque, incluyendo la pseudocápsula tumoral y márgenes libres de tumor, ya que así podríamos lograr un buen resultado local y a largo plazo.
A nuestro paciente se le realizó:
1.- exéresis transpedicular del tumor de escápula derecha; y
2.- la exéresis del tumor sésil localizado en metáfisis de húmero izquierdo, cuya rotura era la responsable del dolor que presentaba en hombro izquierdo.
Los osteocondromas son tumores benignos y no afectan la esperanza de vida (3).
El riesgo de malignización hacia osteosarcoma es aproximadamente del 1% y, en caso de producirse, el pronóstico dependerá del grado histológico, siendo:
a.- la supervivencia a los 10 años del 83% para el grado I y
b.- del 29% para el grado III (3).
{kind=link}
7. Bibliografía:
1. Gupta PP, Agarwal D. A middle-aged manwith persisting chest opacity and permanent bony swelling. CMAJ 2006 Nov 15 ; 31 (24): E920-4
2. Manish Chadha, MS; Arun Pal Singh, MS. Secondary chondrosarcoma of the cuboid bone in a patient with multiple exostosis. Can J Surg, 2008.Vol. 51 (1).
3. Judith VMG Bovée. Multiple osteochondromas. Orphanet Journal of Rare Diseases 2008, 3:3
4. Carolyn M. Sofka, MD& Gregory R. Saboeiro, MD& Robert Schneider, MD. Multiple Hereditary Exostoses. HSSJ (2005) 1:49–51.
5. J V M G Bovée, R J B Sakkers, M J A Geirnaerdt, A H M Taminiau, P C W Hogendoorn. Intermediate grade osteosarcoma and chondrosarcoma
6. Arising in an osteochondroma. A case report of a patient with hereditary multiple exostoses.J Clin Pathol 2002;55:226–229
Fuente: Portalesmadicos.com
Dolor de hombro en paciente con exostosis multiple. A proposito de un caso clinico
Blogalaxia Tags: medicina medicina alternativa tratamientos naturales remedios caseros remedios para
Webs Recomendadas: remedios y tratamientos naturalesmedicina natural y tratamientostratamientos naturalesTratamientos Medicinales Gratis
viernes, 6 de agosto de 2010
Una pareja de padres negros tiene un bebé blanco
http://www.20minutos.es/noticia/770430/bebe/blanco/negros/
Una pareja de padres negros tiene un bebé blanco en contra de las leyes de la genética
El bebé es una niña rubia que según los expertos no es albina, lo que les ha desconcertado aún más.
Los padres no dudan de que es su hija, pero no entienden por qué es rubia con los ojos azules.
Los expertos creen que se ha producido una mutución genética desconocida, ya que no tienen antecedentes de mezcla de raza.
Minuteca todo sobre: genética
20MINUTOS.ES. 20.07.2010
Los padres, Ben y Angela, de origen nigeriano pero instalados en Gran Bretaña, ambos de raza negra, no esconden su sorpresa ante el extraño acontecimiento, su bebé es una niña rubia, blanca. Pese a ello, no dudan de que es su hija.
Mientras, los expertos, todavía contrariados, han descartado que sea una niña albina y dicen que lo sucedido va en contra de la genética, según recoge el diario sensacionalista The Sun.
Es hermosa, un bebé milagro, pero aún así, ¿qué diablos pasó aquí?
La madre no lo duda, "es hermosa, un bebé milagro" dice, mientras el padre admite su sorpresa y aunque asegura que está convencido de que es su hija, admite que bromeó con su mujer:
"¿Ella es mía?".
De nombre la han puesto Nmachi, que significa 'la belleza de dios', y es que esta pequeña, rubia y con unos ojos grandes y azules, parece tener una estrecha relación con lo divino, al menos, si se la compara con sus dos hermanos mayores, ambos de color, como sus padres, se podría decir que en su nacimiento se ha obrado un milagro.
Los expertos, desconcertados
Los más desconcertados son los expertos que, tras averiguar que los padres, de origen nigeriano, no tiene ningún tipo de ascendencia en la que haya habido una mezcla de razas, aseguran que el nacimiento de una niña como Nmachi va contra las leyes de la genética.
La amo, su color no importa
Tras descartar que la niña sea albina o que la mujer le haya sido infiel a su pareja, la conclusión de los expertos es que se trata de alguna mutación genética desconocida.
Hasta que la ciencia resuelva este misterio de la genética, lo importante es que la niña tiene una familia que, aunque tiene un color de piel diferente, la ha acogido con un amor incondicional.
"Ella es hermosa y la amo, su color no importa.
Ella es una bebé milagro, pero aún así, ¿qué diablos pasó aquí?" comenta Angela, la madre
Una pareja de padres negros tiene un bebé blanco en contra de las leyes de la genética
El bebé es una niña rubia que según los expertos no es albina, lo que les ha desconcertado aún más.
Los padres no dudan de que es su hija, pero no entienden por qué es rubia con los ojos azules.
Los expertos creen que se ha producido una mutución genética desconocida, ya que no tienen antecedentes de mezcla de raza.
Minuteca todo sobre: genética
20MINUTOS.ES. 20.07.2010
Los padres, Ben y Angela, de origen nigeriano pero instalados en Gran Bretaña, ambos de raza negra, no esconden su sorpresa ante el extraño acontecimiento, su bebé es una niña rubia, blanca. Pese a ello, no dudan de que es su hija.
Mientras, los expertos, todavía contrariados, han descartado que sea una niña albina y dicen que lo sucedido va en contra de la genética, según recoge el diario sensacionalista The Sun.
Es hermosa, un bebé milagro, pero aún así, ¿qué diablos pasó aquí?
La madre no lo duda, "es hermosa, un bebé milagro" dice, mientras el padre admite su sorpresa y aunque asegura que está convencido de que es su hija, admite que bromeó con su mujer:
"¿Ella es mía?".
De nombre la han puesto Nmachi, que significa 'la belleza de dios', y es que esta pequeña, rubia y con unos ojos grandes y azules, parece tener una estrecha relación con lo divino, al menos, si se la compara con sus dos hermanos mayores, ambos de color, como sus padres, se podría decir que en su nacimiento se ha obrado un milagro.
Los expertos, desconcertados
Los más desconcertados son los expertos que, tras averiguar que los padres, de origen nigeriano, no tiene ningún tipo de ascendencia en la que haya habido una mezcla de razas, aseguran que el nacimiento de una niña como Nmachi va contra las leyes de la genética.
La amo, su color no importa
Tras descartar que la niña sea albina o que la mujer le haya sido infiel a su pareja, la conclusión de los expertos es que se trata de alguna mutación genética desconocida.
Hasta que la ciencia resuelva este misterio de la genética, lo importante es que la niña tiene una familia que, aunque tiene un color de piel diferente, la ha acogido con un amor incondicional.
"Ella es hermosa y la amo, su color no importa.
Ella es una bebé milagro, pero aún así, ¿qué diablos pasó aquí?" comenta Angela, la madre
Metacondromatosis
http://orphanet.orpha.net/consor4.01/www/cgi-bin/OC_Exp.php?lng=ES&Expert=2499
Metacondromatosis
Número de Orphanet: ORPHA2499
Prevalencia de enfermedades raras: <1 / 1 000 000
Herencia: Autosómico dominate
Edad de inicio o aparición: Infancia
Código ICD 10: Q78.8
número MIM: 156250
Resumen
La metacondromatosis es un trastorno raro caracterizado por la presencia de múltiples encondromas y lesiones similares a osteocondroma.
Hasta la fecha, se han informado alrededor de 25 casos. La prevalencia es desconocida.
Los primeros signos de metacondromatosis ocurren durante la primera década de la vida.
Con más frecuencia, los osteocondromas ocurren en manos y pies, predominantemente en los dedos, y los encondromas implican las crestas iliacas y la metáfisis de huesos largos.
Los osteocondromas son pequeños y apuntan hacia la placa de crecimiento adyacente.
La metacondromatosis no conduce al acortamiento o arqueamiento de los huesos largos, deformidad de articulaciones o subluxación.
Las lesiones reducen su tamaño o desaparecen espontáneamente.
La metacondromatosis se hereda de manera autosómica dominante, pero el gen responsable y su patogénesis son aún desconocidos.
El diagnóstico se basa en signos clínicos, hallazgos radiográficos y la historia familiar.
Las características radiográficas son osteocondromas en las metáfisis de los huesos tubulares cortos (manos y pies) que apuntan hacia las articulaciones y que coexisten con encondromas.
El diagnóstico diferencial debe incluir osteocondromas múltiples hereditarios en los que los huesos largos están predominantemente afectados y las lesiones apuntan en dirección contraria de la articulación/placa de crecimiento y pueden dar como resultado el acortamiento o deformidad de los huesos afectados.
Otras enfermedades a ser consideradas incluyen
1.- la enfermedad de Ollier no hereditaria y
2.-el síndrome de Maffucci (en la que los encondromas múltiples se encuentran en la médula del hueso y son predominantemente unilaterales), y
3.- displasia epifisaria hemimélica (DEH; caracterizada por un sobrecrecimiento cartilaginoso principalemente localizado en las extremidades inferiores de un lado del cuerpo)
El asesoramiento genético puede ser propuesto a los pacientes y sus familias.
El diagnóstico prenatal no es posible.
Puede considerarse la intervención quirúrgica para eliminar los osteocondromas en los casos de un mal alineamiento grave de dedos de manos y pies.
Hasta ahora no se han detectado casos de transformación neoplásica.
El curso clínico es impredecible ya que puede haber crecimiento simultáneo de algunas lesiones y regresión de otras.
Pueden ocurrir parálisis nerviosa o complicaciones vasculares (necrosis vascular de la cabeza del fémur; consulte este término).
*Autores: Dr. C.M.A. Reijnders y Dr. J.V.M.G. Bovée (Febrero 2009)*.
Metacondromatosis
Número de Orphanet: ORPHA2499
Prevalencia de enfermedades raras: <1 / 1 000 000
Herencia: Autosómico dominate
Edad de inicio o aparición: Infancia
Código ICD 10: Q78.8
número MIM: 156250
Resumen
La metacondromatosis es un trastorno raro caracterizado por la presencia de múltiples encondromas y lesiones similares a osteocondroma.
Hasta la fecha, se han informado alrededor de 25 casos. La prevalencia es desconocida.
Los primeros signos de metacondromatosis ocurren durante la primera década de la vida.
Con más frecuencia, los osteocondromas ocurren en manos y pies, predominantemente en los dedos, y los encondromas implican las crestas iliacas y la metáfisis de huesos largos.
Los osteocondromas son pequeños y apuntan hacia la placa de crecimiento adyacente.
La metacondromatosis no conduce al acortamiento o arqueamiento de los huesos largos, deformidad de articulaciones o subluxación.
Las lesiones reducen su tamaño o desaparecen espontáneamente.
La metacondromatosis se hereda de manera autosómica dominante, pero el gen responsable y su patogénesis son aún desconocidos.
El diagnóstico se basa en signos clínicos, hallazgos radiográficos y la historia familiar.
Las características radiográficas son osteocondromas en las metáfisis de los huesos tubulares cortos (manos y pies) que apuntan hacia las articulaciones y que coexisten con encondromas.
El diagnóstico diferencial debe incluir osteocondromas múltiples hereditarios en los que los huesos largos están predominantemente afectados y las lesiones apuntan en dirección contraria de la articulación/placa de crecimiento y pueden dar como resultado el acortamiento o deformidad de los huesos afectados.
Otras enfermedades a ser consideradas incluyen
1.- la enfermedad de Ollier no hereditaria y
2.-el síndrome de Maffucci (en la que los encondromas múltiples se encuentran en la médula del hueso y son predominantemente unilaterales), y
3.- displasia epifisaria hemimélica (DEH; caracterizada por un sobrecrecimiento cartilaginoso principalemente localizado en las extremidades inferiores de un lado del cuerpo)
El asesoramiento genético puede ser propuesto a los pacientes y sus familias.
El diagnóstico prenatal no es posible.
Puede considerarse la intervención quirúrgica para eliminar los osteocondromas en los casos de un mal alineamiento grave de dedos de manos y pies.
Hasta ahora no se han detectado casos de transformación neoplásica.
El curso clínico es impredecible ya que puede haber crecimiento simultáneo de algunas lesiones y regresión de otras.
Pueden ocurrir parálisis nerviosa o complicaciones vasculares (necrosis vascular de la cabeza del fémur; consulte este término).
*Autores: Dr. C.M.A. Reijnders y Dr. J.V.M.G. Bovée (Febrero 2009)*.
lunes, 2 de agosto de 2010
Las técnicas de secuenciación genética revolucionan la biología
http://www.elperiodico.com/es/noticias/ciencia-y-tecnologia/20100802/las-tecnicas-secuenciacion-genetica-revolucionan-biologia/416003.shtml
CIENCIA
Las técnicas de secuenciación genética revolucionan la biología
Un análisis es 50.000 veces más rápido y 10.000 más barato que hace solo una década
Hace 25 años que en España se descifraron los primeros genes de plantas y animales
Lunes, 2 de agosto del 2010 /MICHELE CATANZARO / Barcelona
La secuenciación de genes, una técnica de análisis que permite descifrar el mensaje contenido en el ADN, la molécula fundamental de la vida, se ha convertido en una técnica omnipresente en los laboratorios:
a.- los médicos la usan para diagnosticar enfermedades monogenéticas;
b.- los paleontólogos, para esbozar el retrato del hombre de neandertal, e incluso
c.- los policías se benefician de ella para identificar a víctimas y criminales.
Pero no siempre ha sido así. Y mucho menos en España.
«Hace 3 décadas hubiera sido impensable. Para aislar y secuenciar 2 genes necesitamos 2 años», recuerda Salomé Prat, que en 1985, cuando era doctoranda en el Institut de Biologia de Barcelona, descifró los primeros genes de plantas en España.
El mismo año, en Madrid, el equipo de Joan Modolell y Margarita Salas, del Centro de Biología Molecular Severo Ochoa, secuenció genes del primer animal, la mosca de la fruta.
En la última década, los costes de la secuenciación se han reducido en 10.000 veces y la velocidad ha aumentado en 50.000, según datos publicados en abril por la revista Nature. «Actualmente, los individuos cuyo ADN se conoce con todo detalle son unas pocas decenas, pero todo apunta a que esta cifra crecerá rápidamente», pone como ejemplo Pere Puigdomènech, director del Centre de Recerca en Agrigenòmica de Barcelona, que codirigió la tesis de Salomé Prat junto con el fallecido Jordi Cortadas.
EQUIPO LOCAL.
«El cambio responde a unos métodos cada vez más eficientes», explica Prat, que actualmente es investigadora del Centro Nacional de Biotecnología (CNB), en Madrid.
A principios de los 80, en España solo se habían conseguido secuencias de algunos virus en Madrid.
En su opinión, un avance muy importante se produjo en 1982 cuando se organizó en Barcelona un curso en el que investigadores internacionales transfirieron a la comunidad local las técnicas más modernas.
«Entonces ya estaba claro que la secuenciación iba a ser una pieza clave de la ciencia y la tecnología alimentaria», explica Prat.
Aplicando esta técnica a las plantas, los investigadores esperaban tanto entender su funcionamiento como aprender a modificarlas.
«El maíz es deficiente en ciertos aminoácidos esenciales: si se pudiera aumentar su expresión, las dietas basadas casi exclusivamente en maíz que se dan en ciertas partes del mundo serían más nutritivas», comenta.
En 1985, Prat tardó 2 años en aislar y secuenciar 2 genes de maíz que contenían menos de 1.000 pares de bases, las letras que componen el ADN.
En el 2010, en cambio, el equipo de Puigdomènech ha tardado 6 meses en secuenciar el genoma completo del melón, formado por 480 millones de pares.
El salto de calidad tiene mucho que ver con la tecnología.
Prat y Puigdomènech recuerdan que en los 80 se utilizaban métodos químicos que requerían un engorroso proceso de purificación del ADN.
Además, los genes extraídos de las plantas se tenían que insertar en bacterias que los «amplificaban», es decir, producían muchas copias.
En los años 90, las bacterias fueron sustituidas por la llamada técnica de la PCR, que utiliza enzimas, y los métodos de secuenciación antiguos dejaron paso a métodos masivos que permiten hacer secuencias en paralelo.
Sin embargo, los primeros estudios no fueron en absoluto inútiles.
«De esos estudios surgió una empresa, Era Biotech, que se halla en el Parc Científic de Barcelona –señala Puigdomènech–. Esa spin-off comercializa una técnica para almacenar proteínas en células vegetales y animales».
El investigador subraya que entonces los investigadores españoles se colocaron en la vanguardia de la ciencia europea, pero alerta de que los recortes en la inversión pública en ciencia podrían devolverlos a una posición secundaria.
Prat, por su parte, apunta a la rechazo de los transgénicos como un factor que limita la inversión en este campo.
CIENCIA
Las técnicas de secuenciación genética revolucionan la biología
Un análisis es 50.000 veces más rápido y 10.000 más barato que hace solo una década
Hace 25 años que en España se descifraron los primeros genes de plantas y animales
Lunes, 2 de agosto del 2010 /MICHELE CATANZARO / Barcelona
La secuenciación de genes, una técnica de análisis que permite descifrar el mensaje contenido en el ADN, la molécula fundamental de la vida, se ha convertido en una técnica omnipresente en los laboratorios:
a.- los médicos la usan para diagnosticar enfermedades monogenéticas;
b.- los paleontólogos, para esbozar el retrato del hombre de neandertal, e incluso
c.- los policías se benefician de ella para identificar a víctimas y criminales.
Pero no siempre ha sido así. Y mucho menos en España.
{kind=link}
«Hace 3 décadas hubiera sido impensable. Para aislar y secuenciar 2 genes necesitamos 2 años», recuerda Salomé Prat, que en 1985, cuando era doctoranda en el Institut de Biologia de Barcelona, descifró los primeros genes de plantas en España.
El mismo año, en Madrid, el equipo de Joan Modolell y Margarita Salas, del Centro de Biología Molecular Severo Ochoa, secuenció genes del primer animal, la mosca de la fruta.
En la última década, los costes de la secuenciación se han reducido en 10.000 veces y la velocidad ha aumentado en 50.000, según datos publicados en abril por la revista Nature. «Actualmente, los individuos cuyo ADN se conoce con todo detalle son unas pocas decenas, pero todo apunta a que esta cifra crecerá rápidamente», pone como ejemplo Pere Puigdomènech, director del Centre de Recerca en Agrigenòmica de Barcelona, que codirigió la tesis de Salomé Prat junto con el fallecido Jordi Cortadas.
EQUIPO LOCAL.
«El cambio responde a unos métodos cada vez más eficientes», explica Prat, que actualmente es investigadora del Centro Nacional de Biotecnología (CNB), en Madrid.
A principios de los 80, en España solo se habían conseguido secuencias de algunos virus en Madrid.
En su opinión, un avance muy importante se produjo en 1982 cuando se organizó en Barcelona un curso en el que investigadores internacionales transfirieron a la comunidad local las técnicas más modernas.
«Entonces ya estaba claro que la secuenciación iba a ser una pieza clave de la ciencia y la tecnología alimentaria», explica Prat.
Aplicando esta técnica a las plantas, los investigadores esperaban tanto entender su funcionamiento como aprender a modificarlas.
«El maíz es deficiente en ciertos aminoácidos esenciales: si se pudiera aumentar su expresión, las dietas basadas casi exclusivamente en maíz que se dan en ciertas partes del mundo serían más nutritivas», comenta.
En 1985, Prat tardó 2 años en aislar y secuenciar 2 genes de maíz que contenían menos de 1.000 pares de bases, las letras que componen el ADN.
En el 2010, en cambio, el equipo de Puigdomènech ha tardado 6 meses en secuenciar el genoma completo del melón, formado por 480 millones de pares.
El salto de calidad tiene mucho que ver con la tecnología.
Prat y Puigdomènech recuerdan que en los 80 se utilizaban métodos químicos que requerían un engorroso proceso de purificación del ADN.
Además, los genes extraídos de las plantas se tenían que insertar en bacterias que los «amplificaban», es decir, producían muchas copias.
En los años 90, las bacterias fueron sustituidas por la llamada técnica de la PCR, que utiliza enzimas, y los métodos de secuenciación antiguos dejaron paso a métodos masivos que permiten hacer secuencias en paralelo.
Sin embargo, los primeros estudios no fueron en absoluto inútiles.
«De esos estudios surgió una empresa, Era Biotech, que se halla en el Parc Científic de Barcelona –señala Puigdomènech–. Esa spin-off comercializa una técnica para almacenar proteínas en células vegetales y animales».
El investigador subraya que entonces los investigadores españoles se colocaron en la vanguardia de la ciencia europea, pero alerta de que los recortes en la inversión pública en ciencia podrían devolverlos a una posición secundaria.
Prat, por su parte, apunta a la rechazo de los transgénicos como un factor que limita la inversión en este campo.
domingo, 1 de agosto de 2010
Médicos operan pierna "equivocada" un Osteocondroma
http://www.elmundo.es/elmundo/2010/07/30/madrid/1280476984.html
http://www.reporte360.com/detalle.php?id=42655&c=7
Médicos operan pierna "equivocada"
El cirujano culpa a la paciente en el informe de alta porque le indicó la pierna errónea, situación que niega.
Por su lado, el Defensor del Paciente pide que se abra una investigación para aclarar el hecho.
30 Julio 2010.Caracas, Reporte360 -
Una joven mujer madrileña debía ser operada de la pierna derecha, donde tenía un osteocondroma, un tumor óseo benigno, pero cuando terminó la operación el médico tratante confesó que se había equivocado de pierna, reseñó el diario español Qué!
Horas más tarde, la joven despertó y se dio cuenta que había sido operada de ambas piernas por una equivocación médica.
El rotativo contó que la madre de la paciente le explicó a su hija el inconveniente que surgió en el quirófano.
15 días después de la operación la pierna donde estaba el tumor sana bien.
Sin embargo, su pierna izquierda quedó debilitada, luego de que los médicos la abrieron por confusión.
Según la versión de la familia reseñada por el diario, tras la primera operación fallida el médico se justificó en que los informes estaban mal.
Después de la IIª operación, esta vez en la pierna correcta, el padre de la paciente recuerda que el médico argumentó que los osteocondromas suelen ser bilaterales, por lo que tenían que abrir la pierna izquierda también.
Por otro lado, el Defensor del Paciente instó a la Fiscalía a abrir una investigación, puesto que consideran que la "equivocación es inasumible".
Su presidenta, Carmen Flores, subraya que este tipo de casos suceden "más de lo que deberían", posiblemente por "presión asistencial o porque --los médicos-- van muy deprisa" por falta de recursos en los hospitales.
AF / Reporte360
http://www.reporte360.com/detalle.php?id=42655&c=7
Médicos operan pierna "equivocada"
El cirujano culpa a la paciente en el informe de alta porque le indicó la pierna errónea, situación que niega.
Por su lado, el Defensor del Paciente pide que se abra una investigación para aclarar el hecho.
30 Julio 2010.Caracas, Reporte360 -
Una joven mujer madrileña debía ser operada de la pierna derecha, donde tenía un osteocondroma, un tumor óseo benigno, pero cuando terminó la operación el médico tratante confesó que se había equivocado de pierna, reseñó el diario español Qué!
Horas más tarde, la joven despertó y se dio cuenta que había sido operada de ambas piernas por una equivocación médica.
El rotativo contó que la madre de la paciente le explicó a su hija el inconveniente que surgió en el quirófano.
15 días después de la operación la pierna donde estaba el tumor sana bien.
Sin embargo, su pierna izquierda quedó debilitada, luego de que los médicos la abrieron por confusión.
Según la versión de la familia reseñada por el diario, tras la primera operación fallida el médico se justificó en que los informes estaban mal.
Después de la IIª operación, esta vez en la pierna correcta, el padre de la paciente recuerda que el médico argumentó que los osteocondromas suelen ser bilaterales, por lo que tenían que abrir la pierna izquierda también.
Por otro lado, el Defensor del Paciente instó a la Fiscalía a abrir una investigación, puesto que consideran que la "equivocación es inasumible".
Su presidenta, Carmen Flores, subraya que este tipo de casos suceden "más de lo que deberían", posiblemente por "presión asistencial o porque --los médicos-- van muy deprisa" por falta de recursos en los hospitales.
AF / Reporte360
Suscribirse a:
Comentarios (Atom)